Quickstart¶
This guide will walk you through the basic usage of rna-tools.
Let’s get started with some examples.
fetch a structure from the PDB database¶
Example:
$ rna_pdb_tools.py --fetch 1xjr
downloading...1xjr ok
fetch a biologicaly assembly¶
Example:
$ rna_pdb_tools.py --fetch-ba 1xjr
downloading...1xjr_ba.pdb ok
or over a list of pdb ids in a text file:
$ cat data/pdb_ids.txt
1y26
1fir
$ while read p; do rna_pdb_tools.py --fetch-ba $p; done < data/pdb_ids.txt
downloading...1y26_ba.pdb ok
downloading...1fir_ba.pdb ok
$ ls *.pdb
1fir_ba.pdb 1y26_ba.pdb
get sequences of a bunch of PDB files¶
Example:
$ rna_pdb_tools.py --get-seq *.pdb
# 1xjr
> A:1-47
GGAGUUCACCGAGGCCACGCGGAGUACGAUCGAGGGUACAGUGAAUU
# 6TNA
> A:1-76
GCGGAUUUAgCUCAGuuGGGAGAGCgCCAGAcUgAAgAucUGGAGgUCcUGUGuuCGaUCCACAGAAUUCGCACCA
# rp2_bujnicki_1_rpr
> A:1-15
CCGGAGGAACUACUG
> B:1-10
CCGGCAGCCU
> C:1-15
CCGGAGGAACUACUG
> D:1-10
CCGGCAGCCU
> E:1-15
CCGGAGGAACUACUG
> F:1-10
CCGGCAGCCU
> G:1-15
CCGGAGGAACUACUG
> H:1-10
CCGGCAGCCU
in some more fancy way ;-)
$ rna_pdb_tools.py --get-seq \
--oneline 3_bujnicki_1_rpr* \
--color-seq --compact

get secondary structures of your PDB files¶
Python parser to 3dna <http://x3dna.org/>.
Installation:
# install the code from http://forum.x3dna.org/downloads/3dna-download/
Create a copy of the rna_x3dna_config_local_sample.py (remove "_sample") present in rna-tools/rna_tools/tools/rna_x3dna folder.
Edit this line :
BINARY_PATH = <path to your x3dna-dssr file>
matching the path with the path of your x3dna-dssr file.
e.g. in my case: BINARY_PATH = ~/bin/x3dna-dssr.bin
For one structure you can run this script as:
[mm] py3dna$ git:(master) ✗ ./rna_x3dna.py test_data/1xjr.pdb
test_data/1xjr.pdb
>1xjr nts=47 [1xjr] -- secondary structure derived by DSSR
gGAGUUCACCGAGGCCACGCGGAGUACGAUCGAGGGUACAGUGAAUU
..(((((((...((((.((((.....))..))..))).).)))))))
For multiple structures in the folder, run the script like this:
[mm] py3dna$ git:(master) ✗ ./rna_x3dna.py test_data/*
test_data/1xjr.pdb
>1xjr nts=47 [1xjr] -- secondary structure derived by DSSR
gGAGUUCACCGAGGCCACGCGGAGUACGAUCGAGGGUACAGUGAAUU
..(((((((...((((.((((.....))..))..))).).)))))))
test_data/6TNA.pdb
>6TNA nts=76 [6TNA] -- secondary structure derived by DSSR
GCGGAUUUAgCUCAGuuGGGAGAGCgCCAGAcUgAAgAPcUGGAGgUCcUGUGtPCGaUCCACAGAAUUCGCACCA
(((((((..((((.....[..)))).((((.........)))).....(((((..]....))))))))))))....
test_data/rp2_bujnicki_1_rpr.pdb
>rp2_bujnicki_1_rpr nts=100 [rp2_bujnicki_1_rpr] -- secondary structure derived by DSSR
CCGGAGGAACUACUG&CCGGCAGCCU&CCGGAGGAACUACUG&CCGGCAGCCU&CCGGAGGAACUACUG&CCGGCAGCCU&CCGGAGGAACUACUG&CCGGCAGCCU
[[[[(((.....(((&{{{{))))))&(((((((.....(.(&]]]]).))))&[[[[[[......[[[&))))]]].]]&}}}}(((.....(((&]]]]))))))
Warning
This script should not be used in this given form with Parallel because it process output files from x3dna that are named always in the same way, e.g. dssr-torsions.txt. #TODO
- class rna_tools.tools.rna_x3dna.rna_x3dna.x3DNA(pdbfn, show_log=False, verbose=False)[source]¶
Atributes:
curr_fn report
- get_ion_water_report()[source]¶
@todo File name: /tmp/tmp0pdNHS
no. of DNA/RNA chains: 0 [] no. of nucleotides: 174 no. of waters: 793 no. of metals: 33 [Na=29, Mg=1, K=3]
- get_torsions(outfn) str[source]¶
Get torsion angles into ‘torsion.csv’ file:
nt,id,res,alpha,beta,gamma,delta,epsilon,zeta,e-z,chi,phase-angle,sugar-type,ssZp,Dp,splay,bpseq 1,g,A.GTP1,nan,nan,142.1,89.5,-131.0,-78.3,-53(BI),-178.2(anti),358.6(C2’-exo),~C3’-endo,4.68,4.68,29.98,0 2,G,A.G2,-75.8,-167.0,57.2,79.5,-143.4,-69.7,-74(BI),-169.2(anti),5.8(C3’-endo),~C3’-endo,4.68,4.76,25.61,0
delete a part of of your structure¶
Examples:
$ for i in *pdb; do rna_pdb_tools.py --delete A:46-56 $i > ../rpr_rm_loop/$i ; done
go over all files in the current directory, remove a fragment of chain A, residues between 46-56 (including them) and save outputs to in the folder rpr_rm_loops.
get numbering of your structure and rename chains¶
Rename chain B in structure 4_das_1_rpr.pdb:
$ rna_pdb_tools.py --get-seq 4_das_1_rpr.pdb
> 4_das_1_rpr.pdb B:1-126
GGCUUAUCAAGAGAGGUGGAGGGACUGGCCCGAUGAAACCCGGCAACCACUAGUCUAGCGUCAGCUUCGGCUGACGCUAGGCUAGUGGUGCCAAUUCCUGCAGCGGAAACGUUGAAAGAUGAGCCA
$ rna_pdb_tools.py --edit 'B:1-126>A:1-126' 4_das_1_rpr.pdb > 4_das_1_rpr2.pdb
$ rna_pdb_tools.py --get-seq 4_das_1_rpr2.pdb
> 4_das_1_rpr2.pdb A:1-126
GGCUUAUCAAGAGAGGUGGAGGGACUGGCCCGAUGAAACCCGGCAACCACUAGUCUAGCGUCAGCUUCGGCUGACGCUAGGCUAGUGGUGCCAAUUCCUGCAGCGGAAACGUUGAAAGAUGAGCCA
edit your structure (rename chain)¶
Examples:
$ rna_pdb_tools.py --edit 'A:3-21>A:1-19' 1f27_clean.pdb > 1f27_clean_A1-19.pdb
or even:
$ rna_pdb_tools.py --edit 'A:3-21>A:1-19,B:22-32>B:20-30' 1f27_clean.pdb > 1f27_clean_renumb.pdb
or even, even, do rename X chain to A only for Chen’s pdb structures in the folder, in place (so don’t create a new file):
$ for i in *Chen*; do rna_pdb_tools.py --edit 'X:1-125>A:1-125' $i > ${i}_temp; mv ${i}_temp ${i}; done
# do only edit for Chen's pdb structures, in place.
extract part of your structure¶
Example:
$ rna_pdb_tools.py --extract A:1-4 13_Bujnicki_1_rpr.pdb
REMARK 250 Model edited with rna-tools
REMARK 250 ver 3.1.14
REMARK 250 https://github.com/mmagnus/rna-tools
REMARK 250 Sat May 23 14:54:05 2020
HEADER extract A:1-4
ATOM 1 P G A 1 -16.883 -12.441 8.021 1.00 0.00 P
ATOM 2 OP1 G A 1 -15.777 -12.225 8.969 1.00 0.00 O
ATOM 3 OP2 G A 1 -16.752 -11.535 6.892 1.00 0.00 O
ATOM 4 O5' G A 1 -16.882 -13.822 7.219 1.00 0.00 O
ATOM 5 C5' G A 1 -16.092 -13.871 6.013 1.00 0.00 C
ATOM 6 C4' G A 1 -16.314 -15.160 5.206 1.00 0.00 C
ATOM 7 O4' G A 1 -17.723 -14.932 4.905 1.00 0.00 O
ATOM 8 C3' G A 1 -15.788 -15.216 3.752 1.00 0.00 C
ATOM 9 O3' G A 1 -14.461 -15.860 3.764 1.00 0.00 O
ATOM 10 C2' G A 1 -16.841 -15.946 2.969 1.00 0.00 C
(...)
ATOM 84 O2 U A 4 -14.553 -5.285 -7.938 1.00 0.00 O
ATOM 85 N3 U A 4 -14.077 -5.583 -5.727 1.00 0.00 N
ATOM 86 C4 U A 4 -13.451 -6.130 -4.622 1.00 0.00 C
ATOM 87 O4 U A 4 -13.706 -5.737 -3.486 1.00 0.00 O
ATOM 88 C5 U A 4 -12.494 -7.167 -4.998 1.00 0.00 C
ATOM 89 C6 U A 4 -12.318 -7.489 -6.300 1.00 0.00 C
find missing atoms in my structure¶
Run:
$ rna_pdb_tools.py --get-rnapuzzle-ready input/1_das_1_rpr_fixed.pdb
HEADER Generated with rna-pdb-tools
HEADER ver 91ed4f8-dirty
HEADER https://github.com/mmagnus/rna-pdb-tools
HEADER Sun Mar 5 10:58:07 2017
REMARK 000 Missing atoms:
REMARK 000 + P B <Residue C het= resseq=1 icode= > residue # 1
REMARK 000 + OP1 B <Residue C het= resseq=1 icode= > residue # 1
REMARK 000 + OP2 B <Residue C het= resseq=1 icode= > residue # 1
REMARK 000 + O5' B <Residue C het= resseq=1 icode= > residue # 1
ATOM 1 P C A 1 -16.936 -3.789 68.770 1.00 11.89 P
ATOM 2 OP1 C A 1 -17.105 -3.675 67.302 1.00 14.35 O
ATOM 3 OP2 C A 1 -15.666 -4.265 69.342 1.00 12.68 O
...
mutate residues¶
For example, to replace the first four residues of chain A into adenines and 13th A of chain B, run:
$ rna_pdb_tools.py --mutate 'A:1A+2A+3A+4A,B:13A' \
--inplace output/205d_rmH2o_mutA1234-B1_inplace.pdb
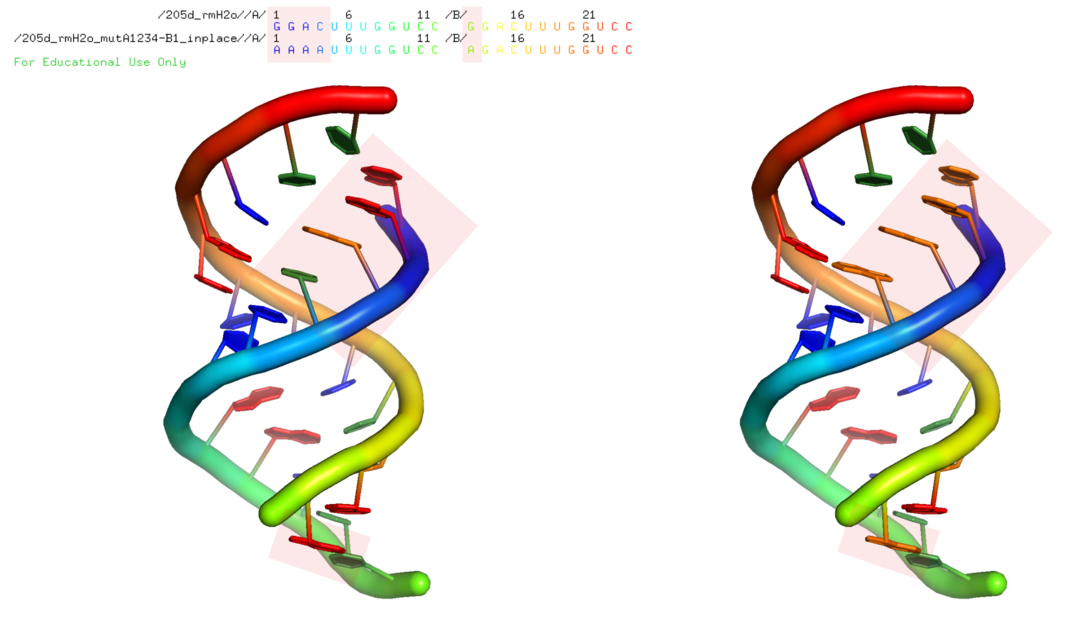
Input structure on the left, mutated structure on the right.
If, for whatever reason, the tool here does not do what you want, use the tool from MC-Fold|MC-Sym Pipeline (go there https://www.major.iric.ca/MC-Pipeline/ and scroll down to the Section: “RNA SEQUENCE MUTATION” at the very bottom of the page).
Moreover, you can also mutate interactively proteins and nucleic acids with PyMOL >2.
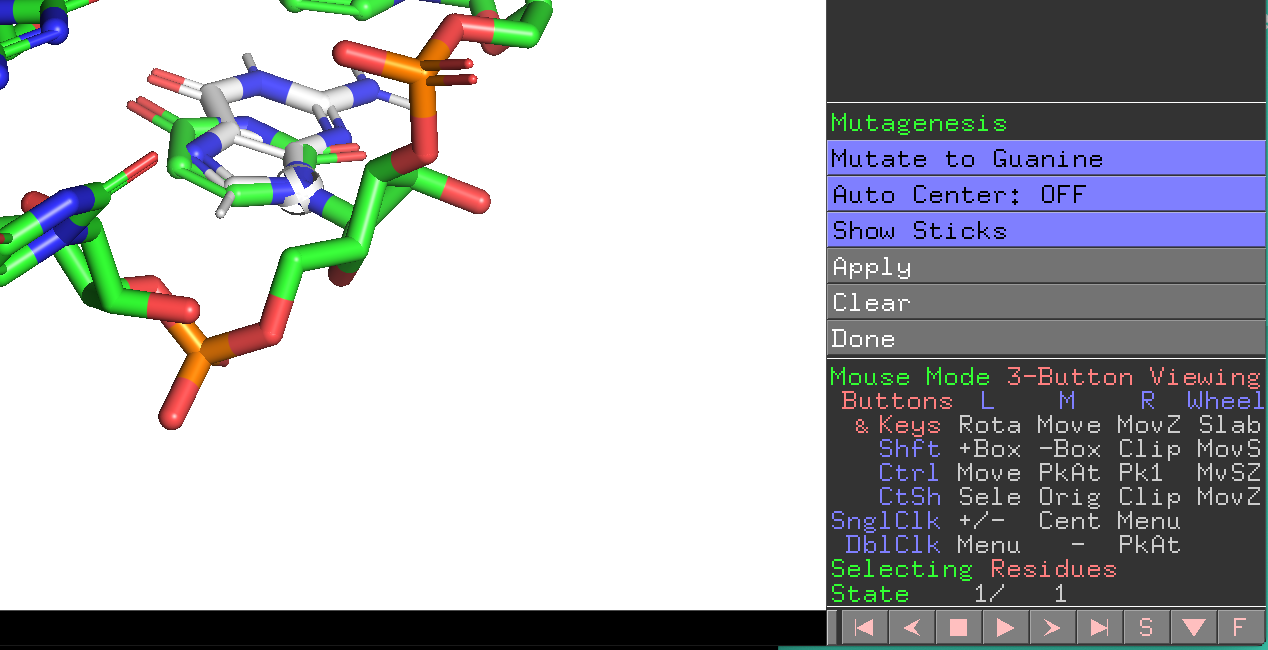
Learn more here https://pymolwiki.org/index.php/Mutagenesis
If you want to mutate with PyMOL with command-line see this https://pymolwiki.org/index.php/Rotkit
add missing atoms¶
The tool is using the function:
- RNAStructure.get_rnapuzzle_ready(renumber_residues=True, fix_missing_atoms=True, rename_chains=True, ignore_op3=False, report_missing_atoms=True, keep_hetatm=False, backbone_only=False, no_backbone=False, bases_only=False, save_single_res=False, ref_frame_only=False, p_only=False, check_geometry=False, conect=False, conect_no_linkage=False, verbose=False)[source]¶
Get rnapuzzle (SimRNA) ready structure.
Clean up a structure, get current order of atoms.
- Parameters:
renumber_residues – boolean, from 1 to …, second chain starts from 1 etc.
fix_missing_atoms – boolean, superimpose motifs from the minilibrary and copy-paste missing atoms, this is super crude, so should be used with caution.
Submission format @http://ahsoka.u-strasbg.fr/rnapuzzles/
Run
rna_tools.rna_tools.lib.RNAStructure.std_resn()before this function to fix names.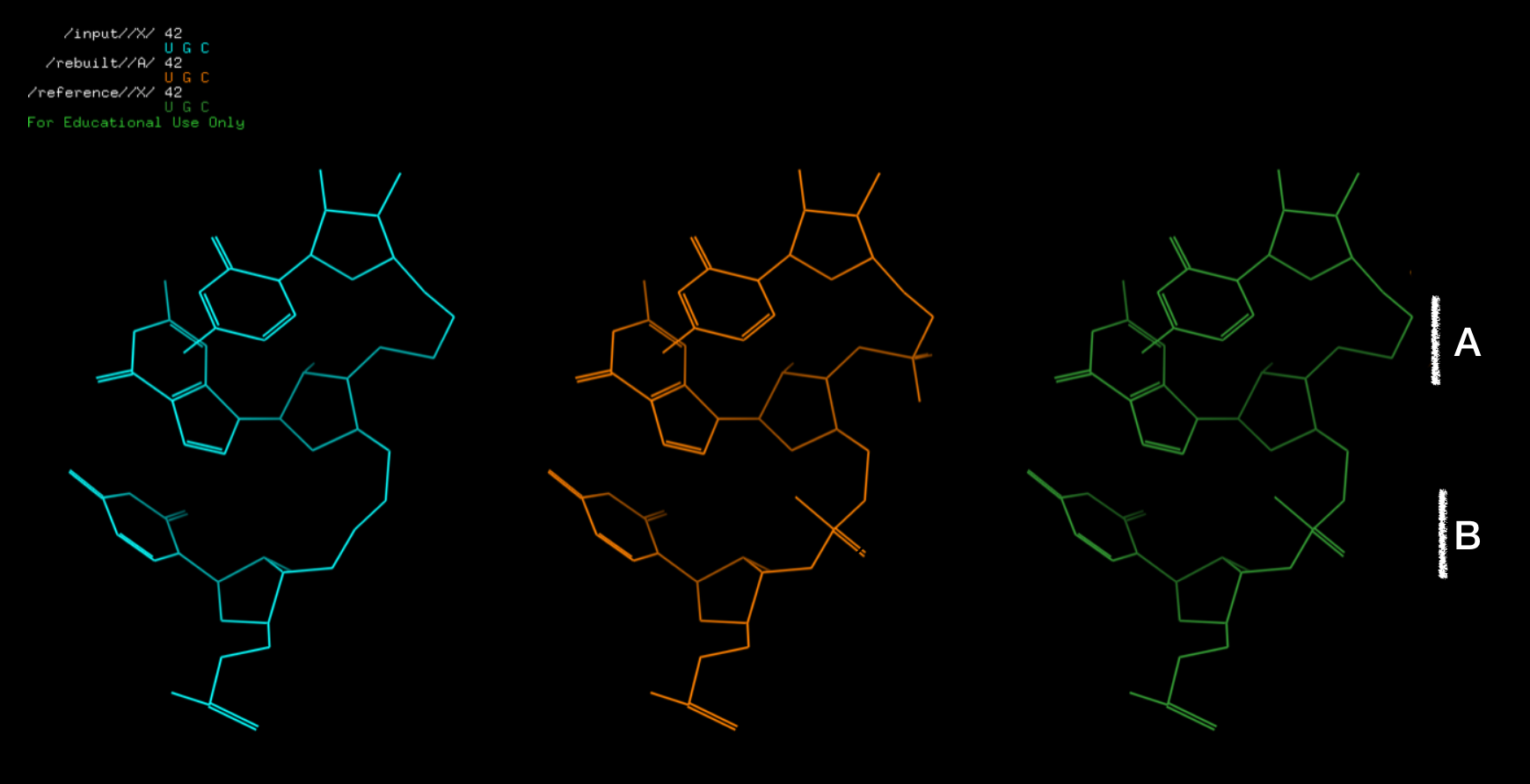
Figure: (Starting from left) input structure, structure with rebuilded atoms, and reference. The B fragment is observed in the reference used here as a “benchmark”, fragment A is reconstructed atoms (not observed in the reference”). 201122
170305 Merged with get_simrna_ready and fixing OP3 terminal added
170308 Fix missing atoms for bases, and O2’
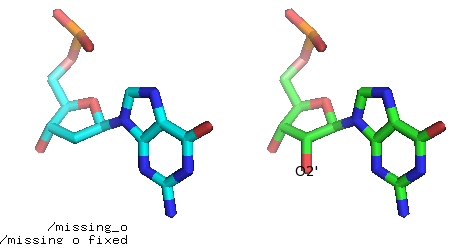
Fig. Add missing O2’ atom (before and after).
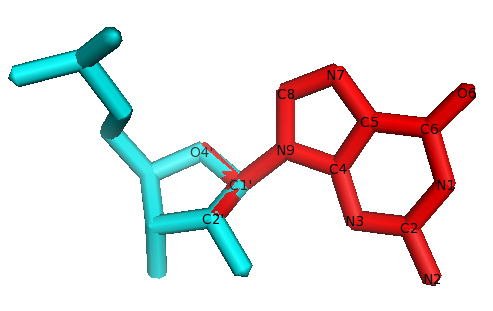
Fig. The residue to fix is in cyan. The G base from the library in red. Atoms O4’, C2’, C1’ are shared between the sugar (in cyan) and the G base from the library (in red). These atoms are used to superimpose the G base on the sugar, and then all atoms from the base are copied to the residues.
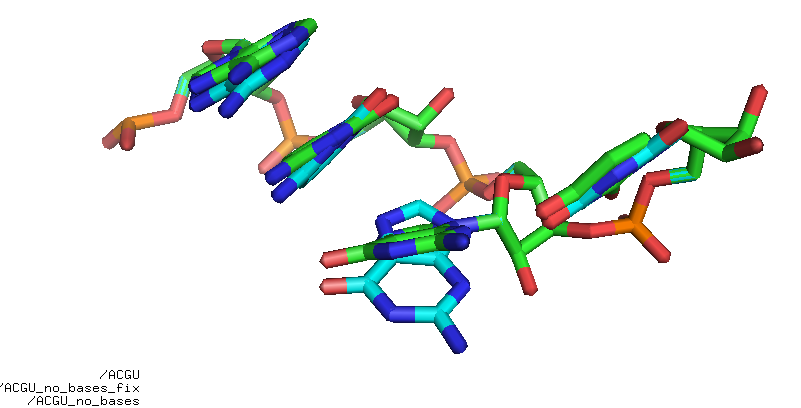
Fig. Rebuild ACGU base-less. It’s not perfect but good enough for some applications.
Warning
It was only tested with the whole base missing!
Warning
requires: Biopython
Selection of atoms:
posphate group (3x, OP1 ,P, OP2),
connector (2x O5’, C5’), /5x
sugar (5x, C4’, O4’, C3’, O3’, C1’, C2’), /10
extra oxygens from sugar (2x, O2’ O3’), for now it’s /12!
A (10x), G (11x), C (8x), U(8x), max 12+11=23
And 27 unique atoms: {‘N9’, ‘O2’, ‘OP2’, “O2’”, “O4’”, ‘C8’, “O3’”, “C1’”, ‘C2’, ‘C6’, “C5’”, ‘N6’, ‘C5’, “C4’”, ‘C4’, “O5’”, “C3’”, ‘O6’, ‘N2’, ‘N7’, ‘OP1’, ‘N1’, ‘N4’, ‘P’, “C2’”, ‘N3’, ‘O4’}.