rna-pdb-tools¶
rna_pdb_tools - a swiss army knife to manipulation of RNA pdb structures
Remove atoms with XYZ equals 0:
(base) ➜ MiniRoseTTA git:(main) ✗ cat 4GXY_min_at0_chemicals.pdb
ATOM 1 OP1 G A 1 50.150 76.113 39.198 1.00 0.00
ATOM 2 P G A 1 50.001 77.254 40.137 1.00 0.00
ATOM 3 OP2 G A 1 48.880 77.258 41.111 1.00 0.00
ATOM 4 O5' G A 1 51.362 77.417 40.948 1.00 0.00
ATOM 5 C5' G A 1 0.000 0.000 0.000 1.00 0.00
ATOM 6 C4' G A 1 0.000 0.000 0.000 1.00 0.00
ATOM 7 O4' G A 1 0.000 0.000 0.000 1.00 0.00
ATOM 8 C3' G A 1 0.000 0.000 0.000 1.00 0.00
to get:
(base) ➜ MiniRoseTTA git:(main) ✗ rna_pdb_tools.py --remove0 4GXY_min_at0_chemicals.pdb
ATOM 1 OP1 G A 1 50.150 76.113 39.198 1.00 0.00
ATOM 2 P G A 1 50.001 77.254 40.137 1.00 0.00
ATOM 3 OP2 G A 1 48.880 77.258 41.111 1.00 0.00
ATOM 4 O5' G A 1 51.362 77.417 40.948 1.00 0.00
ATOM 35 OP1 C A 2 54.648 73.216 44.394 1.00 0.00
ATOM 36 P C A 2 53.712 74.058 43.607 1.00 0.00
ATOM 37 OP2 C A 2 53.842 74.111 42.128 1.00 0.00
ATOM 38 O5' C A 2 52.223 73.613 43.957 1.00 0.00
rna_pdb_tools¶
rna_pdb_tools.py¶
rna_pdb_tools - a swiss army knife to manipulation of RNA pdb structures
Usage:
$ rna_pdb_tools.py --delete A:46-56 --inplace *.pdb
$ rna_pdb_tools.py --get-seq *
# BujnickiLab_RNApuzzle14_n01bound
> A:1-61
# BujnickiLab_RNApuzzle14_n02bound
> A:1-61
CGUUAGCCCAGGAAACUGGGCGGAAGUAAGGCCCAUUGCACUCCGGGCCUGAAGCAACGCG
[...]
See rna_pdb_merge_into_one.py to merge PDB files in the order as you like into one NMR-like (multimodel) file
Examples:
rna_pdb_tools.py --backbone-only --get-rnapuzzle-ready --inplace --suffix=bo examples/4GXY_min.pdb
To extract specific atoms for each residue and write them to separate PDB file (next to the input files, following syntax “<input>_<resid>.pdb”):
rna_pdb_tools.py --rpr input/4GXY_min.pdb --save-single-res --ref-frame-only
Atoms presets:
--backbone-only used only with --get-rnapuzzle-ready, keep only backbone (= remove bases)
--ref-frame-only used only with --get-rnapuzzle-ready, keep only reference frames, OP1 OP2 P
--no-backbone used only with --get-rnapuzzle-ready, remove atoms of backbone (define as P OP1 OP2 O5')
--bases-only used only with --get-rnapuzzle-ready, keep only atoms of bases
-v is for verbose, –version for version ;)
usage: rna_pdb_tools.py [-h] [--version] [-r] [--no-progress-bar]
[--renum-atoms] [--renum-nmr] [--conect]
[--conect-no-linkage] [--renum-residues-dirty]
[--undo] [--delete-anisou] [--remove0] [--fix]
[--to-mol2] [--split-alt-locations] [-c] [--is-pdb]
[--is-nmr] [--nmr-dir NMR_DIR] [--un-nmr] [--orgmode]
[--get-chain GET_CHAIN] [--fetch] [--fetch-cif]
[--fetch-header] [--fetch-ba] [--fetch-chain]
[--fetch-fasta] [--get-seq] [--rgyration]
[--color-seq] [--ignore-files IGNORE_FILES]
[--compact] [--hide-warnings] [--get-ss]
[--rosetta2generic] [--no-hr] [--renumber-residues]
[--dont-rename-chains] [--dont-fix-missing-atoms]
[--inspect] [--collapsed-view] [--cv] [-v]
[--mutate MUTATE] [--edit EDIT]
[--rename-chain RENAME_CHAIN]
[--swap-chains SWAP_CHAINS] [--set-chain SET_CHAIN]
[--replace-chain REPLACE_CHAIN] [--delete DELETE]
[--extract EXTRACT] [--extract-chain EXTRACT_CHAIN]
[--uniq UNIQ] [--chain-first] [--oneline]
[--replace-htm] [--fasta] [--cif2pdb] [--pdb2cif]
[--mdr] [--get-rnapuzzle-ready] [--rpr]
[--keep-hetatm] [--inplace] [--here] [--suffix SUFFIX]
[--replace-hetatm] [--dont-report-missing-atoms]
[--backbone-only] [--p-only] [--ref-frame-only]
[--no-backbone] [--bases-only] [--save-single-res]
file [file ...]
- file¶
file
- -h, --help¶
show this help message and exit
- --version¶
- -r, --report¶
get report
- --no-progress-bar¶
for –no-progress-bar for –rpr
- --renum-atoms¶
renumber atoms, tested with –get-seq
- --renum-nmr¶
- --conect¶
add conect to rpr file
- --conect-no-linkage¶
dont add conect from our residue to another
- --renum-residues-dirty¶
- --undo¶
undo operation of action done –inplace, , rename “backup files” .pdb~ to pdb, ALL files in the folder, not only ~ related to the last action (that you might want to revert, so be careful)
- --delete-anisou¶
remove files with ANISOU records, works with –inplace
- --remove0¶
remove atoms of X=0 position
- --fix¶
fix a PDB file, ! external program, pdbfixer used to fix missing atoms
- --to-mol2¶
fix a PDB file, ! external program, pdbfixer used to fix missing atoms
- --split-alt-locations¶
splits atoms, e.g. for alt locs A and B, it splits atoms two MODELS (all localizations A goes into MODEL1 and all localizations B goes into MODEL2
- -c, --clean¶
get clean structure
- --is-pdb¶
check if a file is in the pdb format
- --is-nmr¶
check if a file is NMR-style multiple model pdb
- --nmr-dir <nmr_dir>¶
make NMR-style multiple model pdb file from a set of files rna_pdb_tools.py –nmr-dir . ‘cwc15_u5_fragments*.pdb’ > ~/Desktop/cwc15-u5.pdb please use ‘’ for pattern file recognition, this is a hack to deal with folders with thousands of models, if you used only *.pdb then the terminal will complain that you selected to many files.
- --un-nmr¶
split NMR-style multiple model pdb files into individual models [biopython], rna_pdb_tools.py –un-nmr split.pdb 2 /Users/magnus/Desktop/3hl2/split_1.pdb /Users/magnus/Desktop/3hl2/split_2.pdb
- --orgmode¶
get a structure in org-mode format <sick!>
- --get-chain <get_chain>¶
get chain, one or many, e.g, A, but now also ABC works
- --fetch¶
fetch file from the PDB db, e.g., 1xjr, use ‘rp’ to fetch, fetch a given join, 4w90:C or 4w90_Cthe RNA-Puzzles standardized_dataset [around 100 MB]
- --fetch-cif¶
- --fetch-header¶
- --fetch-ba¶
fetch biological assembly from the PDB db
- --fetch-chain¶
fetch a structure in extract chain, e.g. 6bk8 H
- --fetch-fasta¶
fetch a fasta/sequence for given PDB ID, e.g. 6bk8
- --get-seq¶
get seq
- --rgyration¶
get seq
- --color-seq¶
color seq, works with –get-seq
- --ignore-files <ignore_files>¶
files to be ingored, .e.g, ‘solution’
- --compact¶
with –get-seq, get it in compact view’ $ rna_pdb_tools.py –get-seq –compact *.pdb # 20_Bujnicki_1 ACCCGCAAGGCCGACGGCGCCGCCGCUGGUGCAAGUCCAGCCACGCUUCGGCGUGGGCGCUCAUGGGU # A:1-68 # 20_Bujnicki_2 ACCCGCAAGGCCGACGGCGCCGCCGCUGGUGCAAGUCCAGCCACGCUUCGGCGUGGGCGCUCAUGGGU # A:1-68 # 20_Bujnicki_3 ACCCGCAAGGCCGACGGCGCCGCCGCUGGUGCAAGUCCAGCCACGCUUCGGCGUGGGCGCUCAUGGGU # A:1-68 # 20_Bujnicki_4
- --hide-warnings¶
hide warnings, works with –get-chain, it hides warnings that given changes are not detected in a PDB file
- --get-ss¶
get secondary structure
- --rosetta2generic¶
convert ROSETTA-like format to a generic pdb
- --no-hr¶
do not insert the header into files
- --renumber-residues¶
by defult is false
- --dont-rename-chains¶
used only with –get-rnapuzzle-ready. By default: –get-rnapuzzle-ready rename chains from ABC.. to stop behavior switch on this option
- --dont-fix-missing-atoms¶
used only with –get-rnapuzzle-ready
- --inspect¶
inspect missing atoms (technically decorator to –get-rnapuzzle-ready without actually doing anything but giving a report on problems)
- --collapsed-view¶
- --cv¶
alias to collapsed_view
- -v, --verbose¶
tell me more what you’re doing, please!
- --mutate <mutate>¶
mutate residues, e.g., –mutate “A:1a+2a+3a+4a,B:1a” to mutate to adenines the first four nucleotides of the chain A and the first nucleotide of the chain B
- --edit <edit>¶
edit ‘A:6>B:200’, ‘A:2-7>B:2-7’
- --rename-chain <rename_chain>¶
edit ‘A>B’ to rename chain A to chain B
- --swap-chains <swap_chains>¶
B>A, rename A to _, then B to A, then _ to B
- --set-chain <set_chain>¶
set chain for all ATOM lines and TER (quite brutal function)
- --replace-chain <replace_chain>¶
a file PDB name with one chain that will be used to replace the chain in the original PDB file, the chain id in this file has to be the same with the chain id of the original chain
- --delete <delete>¶
delete the selected fragment, e.g. A:10-16, or for more than one fragment –delete ‘A:1-25+30-57’
- --extract <extract>¶
extract the selected fragment, e.g. A:10-16, or for more than one fragment –extract ‘A:1-25+30-57’, or even ‘A:1-25+B:30-57’
- --extract-chain <extract_chain>¶
extract chain, e.g. A
- --uniq <uniq>¶
rna_pdb_tools.py –get-seq –uniq ‘[:5]’ –compact –chain-first * | sort A:1-121 ACCUUGCGCAACUGGCGAAUCCUGGGGCUGCCGCCGGCAGUACCC…CA # rp13nc3295_min.out.1 A:1-123 ACCUUGCGCGACUGGCGAAUCCUGAAGCUGCUUUGAGCGGCUUCG…AG # rp13cp0016_min.out.1 A:1-123 ACCUUGCGCGACUGGCGAAUCCUGAAGCUGCUUUGAGCGGCUUCG…AG # zcp_6537608a_ALL-000001_AA A:1-45 57-71 GGGUCGUGACUGGCGAACAGGUGGGAAACCACCGGGGAGCGACCCGCCGCCCGCCUGGGC # solution
- --chain-first¶
- --oneline¶
- --replace-htm¶
- --fasta¶
with –get-seq, show sequences in fasta format, can be combined with –compact (mind, chains will be separated with ‘ ‘ in one line) $ rna_pdb_tools.py –get-seq –fasta –compact input/20_Bujnicki_1.pdb > 20_Bujnicki_1 ACCCGCAAGGCCGACGGC GCCGCCGCUGGUGCAAGUCCAGCCACGCUUCGGCGUGGGCGCUCAUGGGU
- --cif2pdb¶
convert cif to PDB, fancy way
- --pdb2cif¶
[PyMOL Python package required]
- --mdr¶
get structures ready for MD (like rpr but without first)
- --get-rnapuzzle-ready¶
get RNApuzzle ready (keep only standard atoms).’ Be default it does not renumber residues, use –renumber-residues [requires BioPython]
- --rpr¶
alias to get_rnapuzzle ready)
- --keep-hetatm¶
keep hetatoms
- --inplace¶
in place edit the file! [experimental, only for get_rnapuzzle_ready, –delete, –get-ss, –get-seq, –edit-pdb]
- --here¶
save a file next to the original file with auto suffix for –extract it’s .extr.pdb
- --suffix <suffix>¶
when used with –inplace allows you to change a name of a new file, –suffix del will give <file>_del.pdb (mind added _)
- --replace-hetatm¶
replace ‘HETATM’ with ‘ATOM’ [tested only with –get-rnapuzzle-ready]
- --dont-report-missing-atoms¶
used only with –get-rnapuzzle-ready
- --backbone-only¶
used only with –get-rnapuzzle-ready, keep only backbone (= remove bases)
- --p-only¶
used only with –get-rnapuzzle-ready, keep p backbone (= remove bases)
- --ref-frame-only¶
used only with –get-rnapuzzle-ready, keep only reference frames, OP1 OP2 P
- --no-backbone¶
used only with –get-rnapuzzle-ready, remove atoms of backbone (define as P OP1 OP2 O5’)
- --bases-only¶
used only with –get-rnapuzzle-ready, keep only atoms of bases
- --save-single-res¶
used only with –get-rnapuzzle-ready, for each residue create a new pdb output file, you can combine it with –bases-only etc.
get RNAPuzzle ready¶
- class rna_tools.rna_tools_lib.RNAStructure(fn='')[source]¶
RNAStructure - handles an RNA pdb file.
- fn¶
path to the structural file, e.g., “../rna_tools/input/4ts2.pdb”
- Type:
string
- name¶
filename of the structural file, “4ts2.pdb”
- Type:
string
- get_rnapuzzle_ready(renumber_residues=True, fix_missing_atoms=True, rename_chains=True, ignore_op3=False, report_missing_atoms=True, keep_hetatm=False, backbone_only=False, no_backbone=False, bases_only=False, save_single_res=False, ref_frame_only=False, p_only=False, check_geometry=False, conect=False, conect_no_linkage=False, verbose=False)[source]¶
Get rnapuzzle (SimRNA) ready structure.
Clean up a structure, get current order of atoms.
- Parameters:
renumber_residues – boolean, from 1 to …, second chain starts from 1 etc.
fix_missing_atoms – boolean, superimpose motifs from the minilibrary and copy-paste missing atoms, this is super crude, so should be used with caution.
Submission format @http://ahsoka.u-strasbg.fr/rnapuzzles/
Run
rna_tools.rna_tools.lib.RNAStructure.std_resn()before this function to fix names.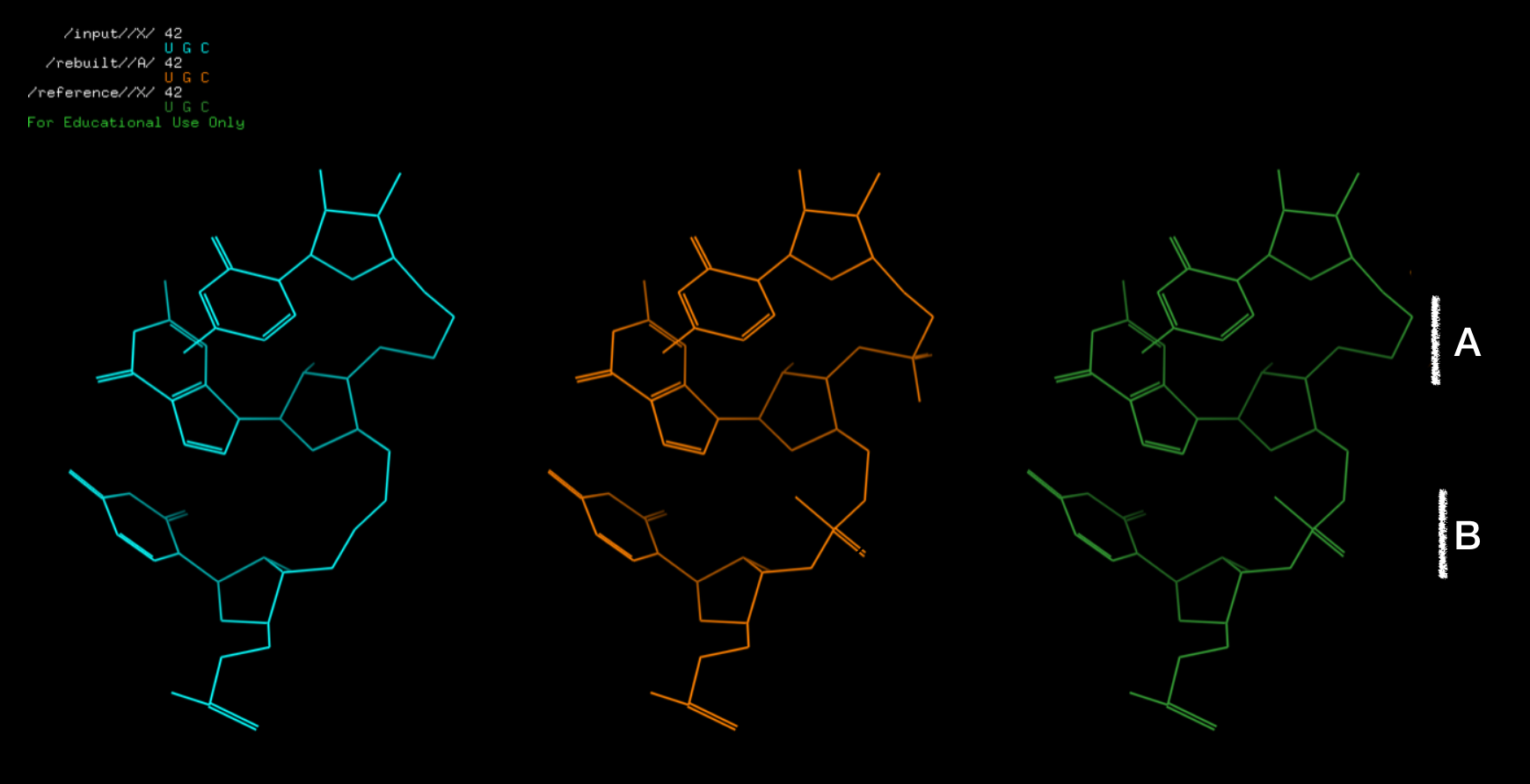
Figure: (Starting from left) input structure, structure with rebuilded atoms, and reference. The B fragment is observed in the reference used here as a “benchmark”, fragment A is reconstructed atoms (not observed in the reference”). 201122
170305 Merged with get_simrna_ready and fixing OP3 terminal added
170308 Fix missing atoms for bases, and O2’
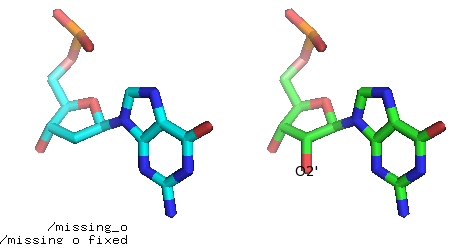
Fig. Add missing O2’ atom (before and after).
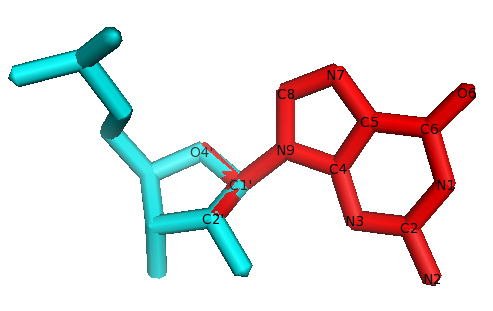
Fig. The residue to fix is in cyan. The G base from the library in red. Atoms O4’, C2’, C1’ are shared between the sugar (in cyan) and the G base from the library (in red). These atoms are used to superimpose the G base on the sugar, and then all atoms from the base are copied to the residues.
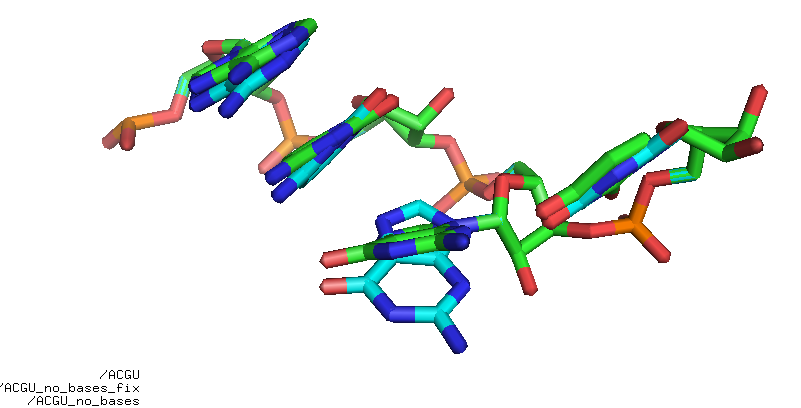
Fig. Rebuild ACGU base-less. It’s not perfect but good enough for some applications.
Warning
It was only tested with the whole base missing!
Warning
requires: Biopython
Selection of atoms:
posphate group (3x, OP1 ,P, OP2),
connector (2x O5’, C5’), /5x
sugar (5x, C4’, O4’, C3’, O3’, C1’, C2’), /10
extra oxygens from sugar (2x, O2’ O3’), for now it’s /12!
A (10x), G (11x), C (8x), U(8x), max 12+11=23
And 27 unique atoms: {‘N9’, ‘O2’, ‘OP2’, “O2’”, “O4’”, ‘C8’, “O3’”, “C1’”, ‘C2’, ‘C6’, “C5’”, ‘N6’, ‘C5’, “C4’”, ‘C4’, “O5’”, “C3’”, ‘O6’, ‘N2’, ‘N7’, ‘OP1’, ‘N1’, ‘N4’, ‘P’, “C2’”, ‘N3’, ‘O4’}.
get sequence¶
Example:
$ rna_pdb_tools.py --get-seq 5_solution_1.pdb
> 5_solution_1.pdb A:1-576
CAUCCGGUAUCCCAAGACAAUCUCGGGUUGGGUUGGGAAGUAUCAUGGCUAAUCACCAUGAUGCAAUCGGGUUGAACACUUAAUUGGGUUAAAACGGUGGGGGACGAUCCCGUAACAUCCGUCCUAACGGCGACAGACUGCACGGCCCUGCCUCAGGUGUGUCCAAUGAACAGUCGUUCCGAAAGGAAG
- class rna_tools.rna_tools_lib.RNAStructure(fn='')[source]¶
RNAStructure - handles an RNA pdb file.
- fn¶
path to the structural file, e.g., “../rna_tools/input/4ts2.pdb”
- Type:
string
- name¶
filename of the structural file, “4ts2.pdb”
- Type:
string
- get_seq(compact=False, chainfirst=False, fasta=False, addfn='', color=False)[source]¶
Get seq (v2) gets segments of chains with correct numbering
Run:
python rna_pdb_seq.py input/1ykq_clx.pdb > 1ykq_clx A:101-111 GGAGCUCGCCC > 1ykq_clx B:201-238 GGGCGAGGCCGUGCCAGCUCUUCGGAGCAAUACUCGGC > 6_solution_0 A:1-19 26-113 117-172 GGCGGCAGGUGCUCCCGACGUCGGGAGUUAAAAGGGAAG
Chains is
{'A': {'header': 'A:1-19 26-113 117-172', 'resi': [1, 2, 3, ..., 19, 26, 27, ..., 172], 'seq': ['G', 'G', 'C', 'G', ... C', 'G', 'U', 'C']}}Chains are in other as the appear in the file.
Warning
take only ATOM and HETATM lines.
fetch¶
Example:
$ rna_pdb_tools.py --fetch 1xjr
downloading...1xjr ok
- rna_tools.rna_tools_lib.fetch(pdb_id, path='.')[source]¶
fetch pdb file from RCSB.org https://files.rcsb.org/download/1Y26.pdb
Args: - pdb_id, but also a chain can be specified, 1jj2:A+B+C
Returns: - a path to a file
TODO: na_pdb_tools.py –extract A:1-25+B:30-57 1jj2.pdb
fetch Biological Assembly¶
Example:
$ rna_pdb_tools.py --fetch-ba 1xjr
downloading...1xjr_ba.pdb ok
or over a list of pdb ids in a text file:
$ cat data/pdb_ids.txt
1y26
1fir
$ while read p; do rna_pdb_tools.py --fetch-ba $p; done < data/pdb_ids.txt
downloading...1y26_ba.pdb ok
downloading...1fir_ba.pdb ok
$ ls *.pdb
1fir_ba.pdb 1y26_ba.pdb
delete¶
Examples:
$ for i in *pdb; do rna_pdb_tools.py --delete A:46-56 $i > ../rpr_rm_loop/$i ; done
go over all files in the current directory, remove a fragment of chain A, residues between 46-56 (including them) and save outputs to in the folder rpr_rm_loops.
edit¶
- rna_tools.rna_tools_lib.edit_pdb(f, args)[source]¶
Edit your structure.
The function can take
A:3-21>A:1-19or even syntax like thisA:3-21>A:1-19,B:22-32>B:20-30and will do an editing.The output is printed, line by line. Only ATOM lines are edited!
Examples:
$ rna_pdb_tools.py --edit 'A:3-21>A:1-19' 1f27_clean.pdb > 1f27_clean_A1-19.pdb
or even:
$ rna_pdb_tools.py --edit 'A:3-21>A:1-19,B:22-32>B:20-30' 1f27_clean.pdb > 1f27_clean_renumb.pdb
RNAStructure (rna_tools_lib)¶
rna_tools_lib.py - main lib file, many tools in this lib is using this file.
- class rna_tools.rna_tools_lib.RNAStructure(fn='')[source]¶
RNAStructure - handles an RNA pdb file.
- fn¶
path to the structural file, e.g., “../rna_tools/input/4ts2.pdb”
- Type:
string
- name¶
filename of the structural file, “4ts2.pdb”
- Type:
string
- check_geometry(verbose=False)[source]¶
Check for correct “Polymer linkage, it should be around 1.6Å with a sigma around 0.01.
Carrascoza, F., Antczak, M., Miao, Z., Westhof, E. & Szachniuk, M. Evaluation of the stereochemical quality of predicted RNA 3D models in the RNA-Puzzles submissions. Rna 28, 250–262 (2022).
Values for 1xjr.pdb:
op.mean(): 1.599316 op.std(): 0.009274074 po.mean(): 1.5984017 po.std(): 0.0069191623requires biopython
- edit_occupancy_of_pdb(pdb, pdb_out, v=False)[source]¶
Make all atoms 1 (flexi) and then set occupancy 0 for seletected atoms. Return False if error. True if OK
- fix_with_qrnas(outfn='', verbose=False)[source]¶
Add missing heavy atom.
A residue is recognized base on a residue names.
Copy QRNAS folder to curr folder, run QRNAS and remove QRNAS.
Warning
QRNAS required (http://genesilico.pl/QRNAS/QRNAS.tgz)
- get_atom_num(line)[source]¶
Extract atom number from a line of PDB file :param * line = ATOM line from a PDB file:
- Output:
atom number (int)
- get_remarks_text()[source]¶
Get remarks as text for given file. This function re-open files, as define as self.fn to get remarks.
Example:
r = RNAStructure(fout) remarks = r.get_remarks_txt() r1 = r.get_res_txt('A', 1) r2 = r.get_res_txt('A', 2) r3 = r.get_res_txt('A', 3) with open(fout, 'w') as f: f.write(remarks) f.write(r1) f.write(r2) f.write(r3) remarks is REMARK 250 Model edited with rna-tools REMARK 250 ver 3.5.4+63.g4338516.dirty REMARK 250 https://github.com/mmagnus/rna-tools REMARK 250 Fri Nov 13 10:15:19 2020
- get_report()[source]¶
- Returns:
report, messages collected on the way of parsing this file
- Return type:
string
- get_res_num(line)[source]¶
Extract residue number from a line of PDB file :param * line = ATOM line from a PDB file:
- Output:
residue number as an integer
- get_res_text(chain_id, resi)[source]¶
Get a residue of given resi of chain_id and return as a text
Example:
r = RNAStructure(fn) print(r.get_res_txt('A', 1)) ATOM 1 O5' G A 1 78.080 -14.909 -0.104 1.00 9.24 O ATOM 2 C5' G A 1 79.070 -15.499 -0.956 1.00 9.70 C ATOM 3 C4' G A 1 78.597 -16.765 -1.648 1.00 9.64 C ATOM 4 O4' G A 1 78.180 -17.761 -0.672 1.00 9.88 O (...)
- get_rnapuzzle_ready(renumber_residues=True, fix_missing_atoms=True, rename_chains=True, ignore_op3=False, report_missing_atoms=True, keep_hetatm=False, backbone_only=False, no_backbone=False, bases_only=False, save_single_res=False, ref_frame_only=False, p_only=False, check_geometry=False, conect=False, conect_no_linkage=False, verbose=False)[source]¶
Get rnapuzzle (SimRNA) ready structure.
Clean up a structure, get current order of atoms.
- Parameters:
renumber_residues – boolean, from 1 to …, second chain starts from 1 etc.
fix_missing_atoms – boolean, superimpose motifs from the minilibrary and copy-paste missing atoms, this is super crude, so should be used with caution.
Submission format @http://ahsoka.u-strasbg.fr/rnapuzzles/
Run
rna_tools.rna_tools.lib.RNAStructure.std_resn()before this function to fix names.Figure: (Starting from left) input structure, structure with rebuilded atoms, and reference. The B fragment is observed in the reference used here as a “benchmark”, fragment A is reconstructed atoms (not observed in the reference”). 201122
170305 Merged with get_simrna_ready and fixing OP3 terminal added
170308 Fix missing atoms for bases, and O2’
Fig. Add missing O2’ atom (before and after).
Fig. The residue to fix is in cyan. The G base from the library in red. Atoms O4’, C2’, C1’ are shared between the sugar (in cyan) and the G base from the library (in red). These atoms are used to superimpose the G base on the sugar, and then all atoms from the base are copied to the residues.
Fig. Rebuild ACGU base-less. It’s not perfect but good enough for some applications.
Warning
It was only tested with the whole base missing!
Warning
requires: Biopython
Selection of atoms:
posphate group (3x, OP1 ,P, OP2),
connector (2x O5’, C5’), /5x
sugar (5x, C4’, O4’, C3’, O3’, C1’, C2’), /10
extra oxygens from sugar (2x, O2’ O3’), for now it’s /12!
A (10x), G (11x), C (8x), U(8x), max 12+11=23
And 27 unique atoms: {‘N9’, ‘O2’, ‘OP2’, “O2’”, “O4’”, ‘C8’, “O3’”, “C1’”, ‘C2’, ‘C6’, “C5’”, ‘N6’, ‘C5’, “C4’”, ‘C4’, “O5’”, “C3’”, ‘O6’, ‘N2’, ‘N7’, ‘OP1’, ‘N1’, ‘N4’, ‘P’, “C2’”, ‘N3’, ‘O4’}.
- get_seq(compact=False, chainfirst=False, fasta=False, addfn='', color=False)[source]¶
Get seq (v2) gets segments of chains with correct numbering
Run:
python rna_pdb_seq.py input/1ykq_clx.pdb > 1ykq_clx A:101-111 GGAGCUCGCCC > 1ykq_clx B:201-238 GGGCGAGGCCGUGCCAGCUCUUCGGAGCAAUACUCGGC > 6_solution_0 A:1-19 26-113 117-172 GGCGGCAGGUGCUCCCGACGUCGGGAGUUAAAAGGGAAGChains is
{'A': {'header': 'A:1-19 26-113 117-172', 'resi': [1, 2, 3, ..., 19, 26, 27, ..., 172], 'seq': ['G', 'G', 'C', 'G', ... C', 'G', 'U', 'C']}}Chains are in other as the appear in the file.
Warning
take only ATOM and HETATM lines.
- is_nmr()[source]¶
True if the file is an NMR-style multiple model pdb
- Returns:
True or Fo
- Return type:
boolean
- is_pdb()[source]¶
Return True if the files is in PDB format.
If self.lines is empty it means that nothing was parsed into the PDB format.
- remove_ion()[source]¶
TER 1025 U A 47 HETATM 1026 MG MG A 101 42.664 34.395 50.249 1.00 70.99 MG HETATM 1027 MG MG A 201 47.865 33.919 48.090 1.00 67.09 MG
- rtype:
object
- set_atom_code(line, code)[source]¶
Add atom name/code:
- ATOM 1 OP2 C A 1 29.615 36.892 42.657 1.00 1.00 O
^^^ ^ and element
- set_res_code(line, code)[source]¶
- Parameters:
lines
code
path (str): The path of the file to wrap field_storage (FileStorage): The :class:Y instance to wrap
temporary (bool): Whether or not to delete the file when the File instance is destructed
- Returns:
A buffered writable file descriptor
- Return type:
BufferedFileStorage
- std_resn()[source]¶
‘Fix’ residue names which means to change them to standard, e.g. RA5 -> A
Works on self.lines, and returns the result to self.lines.
Will change things like:
# URI -> U, URA -> U 1xjr_clx_charmm.pdb:ATOM 101 P URA A 5 58.180 39.153 30.336 1.00 70.94 rp13_Dokholyan_1_URI_CYT_ADE_GUA_hydrogens.pdb:ATOM 82 P URI A 4 501.633 506.561 506.256 1.00 0.00 P
- un_nmr(startwith1=True, directory='', verbose=False)[source]¶
Un NMR - Split NMR-style multiple model pdb files into individual models.
Take self.fn and create new files in the way:
input/1a9l_NMR_1_2_models.pdb input/1a9l_NMR_1_2_models_0.pdb input/1a9l_NMR_1_2_models_1.pdbWarning
This function requires biopython.
rna_pdb_tools.py –un-nmr AF-Q5TCX8-F1-model_v1_core_Ctrim_mdr_MD.pdb
- 36
cat MD_ > md.pdb
- rna_tools.rna_tools_lib.collapsed_view(args)[source]¶
Collapsed view of pdb file. Only lines with C5’ atoms are shown and TER, MODEL, END.
example:
[mm] rna_tools git:(master) $ python rna-pdb-tools.py --cv input/1f27.pdb ATOM 1 C5' A A 3 25.674 19.091 3.459 1.00 16.99 C ATOM 23 C5' C A 4 19.700 19.206 5.034 1.00 12.65 C ATOM 43 C5' C A 5 14.537 16.130 6.444 1.00 8.74 C ATOM 63 C5' G A 6 11.726 11.579 9.544 1.00 9.81 C ATOM 86 C5' U A 7 12.007 7.281 13.726 1.00 11.35 C ATOM 106 C5' C A 8 12.087 6.601 18.999 1.00 12.74 C TER
- rna_tools.rna_tools_lib.edit_pdb(f, args)[source]¶
Edit your structure.
The function can take
A:3-21>A:1-19or even syntax like thisA:3-21>A:1-19,B:22-32>B:20-30and will do an editing.The output is printed, line by line. Only ATOM lines are edited!
Examples:
$ rna_pdb_tools.py --edit 'A:3-21>A:1-19' 1f27_clean.pdb > 1f27_clean_A1-19.pdbor even:
$ rna_pdb_tools.py --edit 'A:3-21>A:1-19,B:22-32>B:20-30' 1f27_clean.pdb > 1f27_clean_renumb.pdb
- rna_tools.rna_tools_lib.fetch(pdb_id, path='.')[source]¶
fetch pdb file from RCSB.org https://files.rcsb.org/download/1Y26.pdb
Args: - pdb_id, but also a chain can be specified, 1jj2:A+B+C
Returns: - a path to a file
TODO: na_pdb_tools.py –extract A:1-25+B:30-57 1jj2.pdb
- rna_tools.rna_tools_lib.fetch_ba(pdb_id, path='.')[source]¶
fetch biological assembly pdb file from RCSB.org
>>> fetch_ba('1xjr') ...
- rna_tools.rna_tools_lib.fetch_cif(cif_id, path='.')[source]¶
fetch biological assembly cif file from RCSB.org
- rna_tools.rna_tools_lib.fetch_cif_ba(cif_id, path='.')[source]¶
fetch biological assembly cif file from RCSB.org
- rna_tools.rna_tools_lib.generate_conect_records(residue_type, shift=0, no_junction=True)[source]¶
Generate CONECT records for an RNA residue (U, A, C, or G).
- rna_tools.rna_tools_lib.load_rnas(path, verbose=True)[source]¶
Load structural files (via glob) and return a list of RNAStructure objects.
Examples:
rnas = rtl.load_rnas('../rna_tools/input/mq/*.pdb')
- rna_tools.rna_tools_lib.replace_atoms(struc_fn, insert_fn, verbose=False)[source]¶
Replace XYZ coordinate of the file (struc_fn) with XYZ from another file (insert_fn).
This can be useful if you want to replace positions of atoms, for example, one base only. The lines are muted based on atom name, residue name, chain, residue index (marked with XXXX below).:
ATOM 11 N1 A 2 27 303.441 273.472 301.457 1.00 0.00 N # file ATOM 1 N1 A 2 27 300.402 273.627 303.188 1.00 99.99 N # insert ATOM 11 N1 A 2 27 300.402 273.627 303.188 1.00 0.00 N # inserted
XXXXXXXXXXXXXXXX # part used to find lines to be replaced
ATOM 1 P A 2 27 295.653 270.783 300.135 1.00119.29 P # next line
- rna_tools.rna_tools_lib.replace_chain(struc_fn, insert_fn, chain_id)[source]¶
Replace chain of the main file (struc_fn) with some new chain (insert_fn) of given chain id.
- rna_tools.rna_tools_lib.set_chain_for_struc(struc_fn, chain_id, save_file_inplace=False, skip_ter=True)[source]¶
Quick & dirty function to set open a fn PDB format, set chain_id and save it to a file. Takes only lines with ATOM and TER.
- rna_tools.rna_tools_lib.sort_strings(l)[source]¶
Sort the given list in the way that humans expect. http://blog.codinghorror.com/sorting-for-humans-natural-sort-order/
Standardize your PDB files¶
rna_standardize.py¶
rna_standardize.py - standardzie RNA PDB structures
Usage:
$ rna_standardize.py <pdb file>
-v is for verbose, –version for version ;)
usage: rna_standardize.py [-h] [--version] [--no-progress-bar] [--quiet]
[--renum-nmr] [--inplace] [-v] [--conect]
[--conect-no-linkage] [--dont-replace-hetatm]
[--keep-hetatm] [--here] [--no-hr] [--cif]
[--check-geometry] [--dont-fix-missing-atoms]
[--mdr] [--renumber-residues] [--suffix SUFFIX]
[--dont-report-missing-atoms] [--dont-rename-chains]
[--backbone-only] [--no-backbone] [--bases-only]
file [file ...]
- file¶
file
- -h, --help¶
show this help message and exit
- --version¶
- --no-progress-bar¶
for –no-progress-bar for –rpr
- --quiet¶
limit prints
- --renum-nmr¶
- --inplace¶
in place edit the file! [experimental, only for get_rnapuzzle_ready, –delete, –get-ss, –get-seq, –edit-pdb]
- -v, --verbose¶
tell me more what you’re doing, please!
- --conect¶
add conect to rpr file
- --conect-no-linkage¶
dont add conect from our residue to another
- --dont-replace-hetatm¶
replace ‘HETATM’ with ‘ATOM’ [tested only with –get-rnapuzzle-ready]
- --keep-hetatm¶
keep hetatoms, [if not replaced anyway with ATOM, see –dont-replace-hetatm
- --here¶
save a file next to the original file with auto suffix for –extract it’s .extr.pdb
- --no-hr¶
do not insert the header into files
- --cif¶
output as cif file
- --check-geometry¶
check connectivity betweeen residues and angles
- --dont-fix-missing-atoms¶
used only with –get-rnapuzzle-ready
- --mdr¶
get structures ready for MD (like rpr but without first)
- --renumber-residues¶
by defult is false
- --suffix <suffix>¶
when used with –inplace allows you to change a name of a new file, –suffix del will give <file>_del.pdb (mind added _)
- --dont-report-missing-atoms¶
used only with –get-rnapuzzle-ready
- --dont-rename-chains¶
used only with –get-rnapuzzle-ready. By default: –get-rnapuzzle-ready rename chains from ABC.. to stop behavior switch on this option
- --backbone-only¶
used only with –get-rnapuzzle-ready, keep only backbone (= remove bases)
- --no-backbone¶
used only with –get-rnapuzzle-ready, remove atoms of backbone (define as P OP1 OP2 O5’)
- --bases-only¶
used only with –get-rnapuzzle-ready, keep only atoms of bases
- class rna_tools.rna_tools_lib.RNAStructure(fn='')[source]¶
RNAStructure - handles an RNA pdb file.
- fn¶
path to the structural file, e.g., “../rna_tools/input/4ts2.pdb”
- Type:
string
- name¶
filename of the structural file, “4ts2.pdb”
- Type:
string
- get_rnapuzzle_ready(renumber_residues=True, fix_missing_atoms=True, rename_chains=True, ignore_op3=False, report_missing_atoms=True, keep_hetatm=False, backbone_only=False, no_backbone=False, bases_only=False, save_single_res=False, ref_frame_only=False, p_only=False, check_geometry=False, conect=False, conect_no_linkage=False, verbose=False)[source]¶
Get rnapuzzle (SimRNA) ready structure.
Clean up a structure, get current order of atoms.
- Parameters:
renumber_residues – boolean, from 1 to …, second chain starts from 1 etc.
fix_missing_atoms – boolean, superimpose motifs from the minilibrary and copy-paste missing atoms, this is super crude, so should be used with caution.
Submission format @http://ahsoka.u-strasbg.fr/rnapuzzles/
Run
rna_tools.rna_tools.lib.RNAStructure.std_resn()before this function to fix names.
Figure: (Starting from left) input structure, structure with rebuilded atoms, and reference. The B fragment is observed in the reference used here as a “benchmark”, fragment A is reconstructed atoms (not observed in the reference”). 201122
170305 Merged with get_simrna_ready and fixing OP3 terminal added
170308 Fix missing atoms for bases, and O2’
Fig. Add missing O2’ atom (before and after).
Fig. The residue to fix is in cyan. The G base from the library in red. Atoms O4’, C2’, C1’ are shared between the sugar (in cyan) and the G base from the library (in red). These atoms are used to superimpose the G base on the sugar, and then all atoms from the base are copied to the residues.
Fig. Rebuild ACGU base-less. It’s not perfect but good enough for some applications.
Warning
It was only tested with the whole base missing!
Warning
requires: Biopython
Selection of atoms:
posphate group (3x, OP1 ,P, OP2),
connector (2x O5’, C5’), /5x
sugar (5x, C4’, O4’, C3’, O3’, C1’, C2’), /10
extra oxygens from sugar (2x, O2’ O3’), for now it’s /12!
A (10x), G (11x), C (8x), U(8x), max 12+11=23
And 27 unique atoms: {‘N9’, ‘O2’, ‘OP2’, “O2’”, “O4’”, ‘C8’, “O3’”, “C1’”, ‘C2’, ‘C6’, “C5’”, ‘N6’, ‘C5’, “C4’”, ‘C4’, “O5’”, “C3’”, ‘O6’, ‘N2’, ‘N7’, ‘OP1’, ‘N1’, ‘N4’, ‘P’, “C2’”, ‘N3’, ‘O4’}.
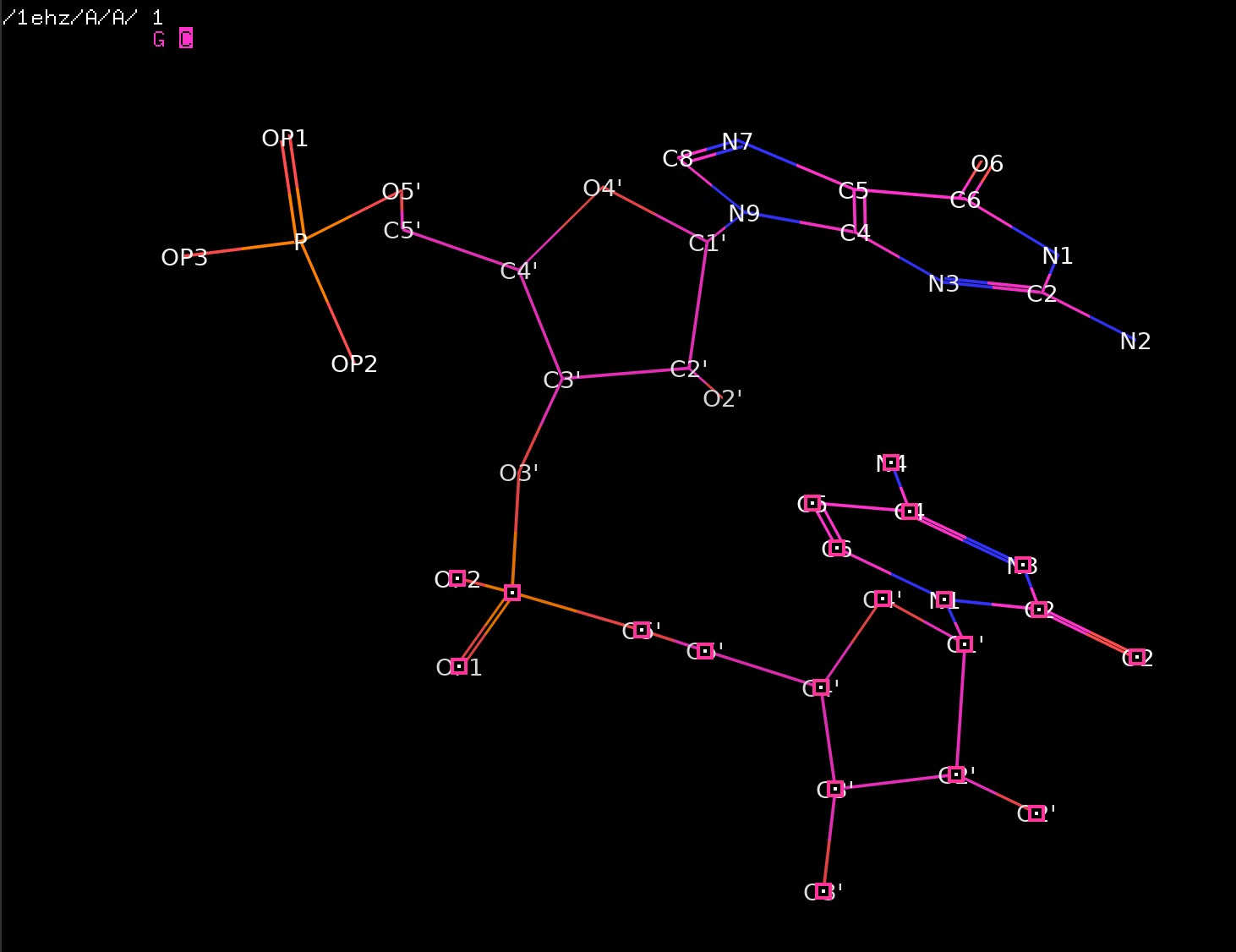
Atoms order¶
Atoms order, A as an example:
ATOM 1 P G A 1 50.626 49.730 50.573 1.00100.19 P
ATOM 2 OP1 G A 1 49.854 48.893 49.562 1.00100.19 O
ATOM 3 OP2 G A 1 52.137 49.542 50.511 1.00 99.21 O
ATOM 4 O5' G A 1 50.161 49.136 52.023 1.00 99.82 O
ATOM 5 C5' G A 1 50.216 49.948 53.210 1.00 98.63 C
ATOM 6 C4' G A 1 50.968 49.231 54.309 1.00 97.84 C
ATOM 7 O4' G A 1 50.450 47.888 54.472 1.00 97.10 O
ATOM 8 C3' G A 1 52.454 49.030 54.074 1.00 98.07 C
ATOM 9 O3' G A 1 53.203 50.177 54.425 1.00 99.39 O
ATOM 10 C2' G A 1 52.781 47.831 54.957 1.00 96.96 C
ATOM 11 O2' G A 1 53.018 48.156 56.313 1.00 96.77 O
ATOM 12 C1' G A 1 51.502 47.007 54.836 1.00 95.70 C
ATOM 13 N9 G A 1 51.628 45.992 53.798 1.00 93.67 N
ATOM 14 C8 G A 1 51.064 46.007 52.547 1.00 92.60 C
ATOM 15 N7 G A 1 51.379 44.966 51.831 1.00 91.19 N
ATOM 16 C5 G A 1 52.197 44.218 52.658 1.00 91.47 C
ATOM 17 C6 G A 1 52.848 42.992 52.425 1.00 90.68 C
ATOM 18 O6 G A 1 52.826 42.291 51.404 1.00 90.38 O
ATOM 19 N1 G A 1 53.588 42.588 53.534 1.00 90.71 N
ATOM 20 C2 G A 1 53.685 43.282 54.716 1.00 91.21 C
ATOM 21 N2 G A 1 54.452 42.733 55.671 1.00 91.23 N
ATOM 22 N3 G A 1 53.077 44.429 54.946 1.00 91.92 N
ATOM 23 C4 G A 1 52.356 44.836 53.879 1.00 92.62 C
ATOM 24 P C A 2 54.635 50.420 53.741 1.00100.19 P
ATOM 25 OP1 C A 2 55.145 51.726 54.238 1.00100.19 O
OP3¶
The first residue will have only OP1 and OP2 (OP3 will be removed):
ATOM 1 OP3 G A 1 50.193 51.190 50.534 1.00 99.85 O
ATOM 2 P G A 1 50.626 49.730 50.573 1.00100.19 P
ATOM 3 OP1 G A 1 49.854 48.893 49.562 1.00100.19 O
ATOM 4 OP2 G A 1 52.137 49.542 50.511 1.00 99.21 O
ATOM 5 O5' G A 1 50.161 49.136 52.023 1.00 99.82 O
ATOM 6 C5' G A 1 50.216 49.948 53.210 1.00 98.63 C
ATOM 7 C4' G A 1 50.968 49.231 54.309 1.00 97.84 C
Listing. An example: 1ehz.pdb.
Torsion angle analysis¶
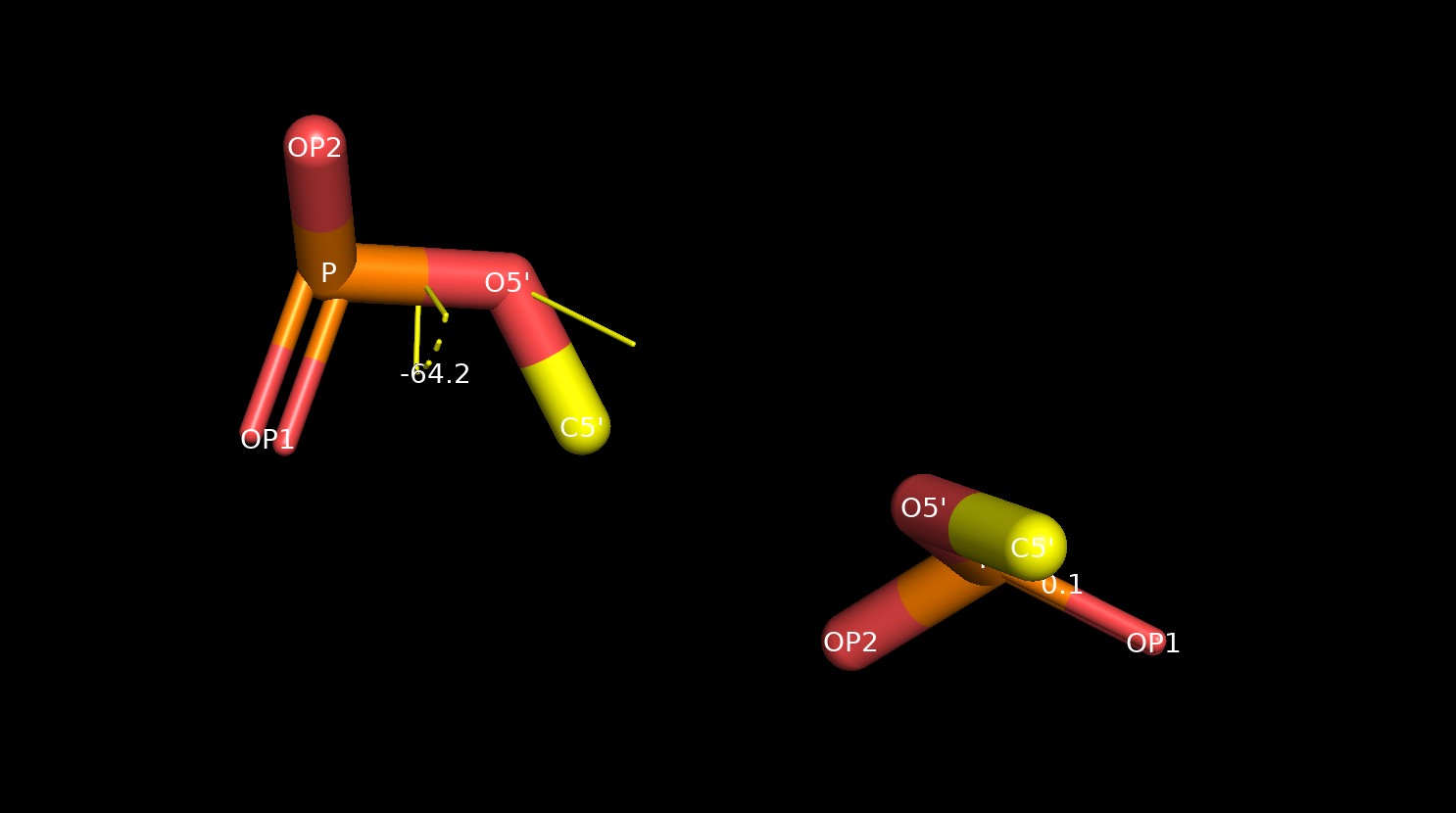
rna_torsions.py¶
Examples:
$ rna_torsions.py ./input/4GXY_min.pdb
f, alphaprime, beta
input ./input/4GXY_min.pdb <Residue G het= resseq=2 icode= >, -64.20924484900823, -143.18546007904766
input ./input/4GXY_min.pdb <Residue C het= resseq=3 icode= >, 2.3394112025736815, 70.4052871669199
Comparison:
$ rna_x3dna.py input/4GXY_min.pdb -s
input: input/4GXY_min.pdb
nt id res alpha beta gamma delta epsilon zeta e-z chi phase-angle sugar-type ssZp Dp splay paired
0 1 G A.G2 NaN -143.2 153.7 82.5 -92.3 -31.9 -60(..) -179.0(anti) 19.5(C3'-endo) ~C3'-endo 4.39 4.56 18.32 no paired
1 2 C A.C3 -111.4 70.4 160.0 80.6 NaN NaN NaN -177.6(anti) 11.1(C3'-endo) ~C3'-endo NaN NaN NaN no paired
usage: rna_torsions.py [-h] [-v] file
- file¶
- -h, --help¶
show this help message and exit
- -v, --verbose¶
be verbose
PDB Edit Bfactor/Occupancy¶
rna_pdb_edit_occupancy_bfactor.py¶
rna_pdb_edit_occupancy_bfactor.py - edit occupancy or bfactor in PDB file.
Example:
rna_pdb_edit_occupancy_bfactor.py --occupancy --select A:1-40,B:1-22 \
--set-to 0 \
19_Bujnicki_Human_4_rpr_n0-000001.pdb
rna_pdb_edit_occupancy_bfactor.py --occupancy \
--select A:1-2 \
--select-atoms P+C4\' \
--set-to 10 \
-o test_data/3w3s_homologymodel_out.PD
--set-not-selected-to 8
test_data/3w3s_homologymodel.pdb
usage: rna_pdb_edit_occupancy_bfactor.py [-h] (--bfactor | --occupancy)
[--select SELECT] [--set-to SET_TO]
[--set-not-selected-to SET_NOT_SELECTED_TO]
[-o OUTPUT] [--verbose]
[--select-atoms SELECT_ATOMS]
file
- file¶
file
- -h, --help¶
show this help message and exit
- --bfactor¶
set bfactor
- --occupancy¶
set occupancy
- --select <select>¶
get chain, e.g A:1-10, works also for multiple chainse.g A:1-40,B:1-22
- --set-to <set_to>¶
set value to, default is 1
- --set-not-selected-to <set_not_selected_to>¶
set value to, default is 0
- -o <output>, --output <output>¶
file output
- --verbose¶
be verbose
- --select-atoms <select_atoms>¶
select only given atomscan be only one atom, e.g. Por more, use ' for prims, e.g. P+C4'
- rna_tools.tools.rna_pdb_edit_occupancy_bfactor.rna_pdb_edit_occupancy_bfactor.edit_occupancy_of_pdb(txt, pdb, pdb_out, bfactor, occupancy, set_to, set_not_selected_to, select_atoms, v=False)[source]¶
Change ouccupancy or bfactor of pdb file.
Load the structure, and first set everything to be set_not_selected_to and then set selected to sel_to.
- Parameters:
txt (str) – A:1-10, selection, what to change
pdb (str) – filename to read as an input
pdb_out (str) – filename to save an output
bfactor (bool) – if edit bfactor
occupancy (bool) – if edit occupancy
set_to (float) – set to this value, if within selection
set_not_selected_to (float) – set to this value, if not within selection
select_atoms (str) – P, P+C4', use + as a separator
v (bool) – be verbose
- Returns:
if OK, save an output to pdb_out
- Return type:
Warning
this function requires BioPython
Add chain to a file¶
Example:
./rna_add_chain.py -c X ../../input/1msy_rnakbmd_decoy999_clx_noChain.pdb > ../../output/1msy_rnakbmd_decoy999_clx_noChain_Xchain.pdb
From:
ATOM 1 O5' U 1 42.778 25.208 46.287 1.00 0.00
ATOM 2 C5' U 1 42.780 26.630 45.876 1.00 0.00
ATOM 3 C4' U 1 42.080 27.526 46.956 1.00 0.00
ATOM 4 O4' U 1 43.013 28.044 47.963 1.00 0.00
ATOM 5 C1' U 1 42.706 29.395 48.257 1.00 0.00
ATOM 6 N1 U 1 43.857 30.305 47.703 1.00 0.00
ATOM 7 C6 U 1 45.057 29.857 47.308 1.00 0.00
ATOM 8 C5 U 1 46.025 30.676 46.763 1.00 0.00
ATOM 9 C4 U 1 45.720 32.110 46.702 1.00 0.00
ATOM 10 O4 U 1 46.444 32.975 46.256 1.00 0.00
to:
ATOM 1 O5' U X 1 42.778 25.208 46.287 1.00 0.00
ATOM 2 C5' U X 1 42.780 26.630 45.876 1.00 0.00
ATOM 3 C4' U X 1 42.080 27.526 46.956 1.00 0.00
ATOM 4 O4' U X 1 43.013 28.044 47.963 1.00 0.00
ATOM 5 C1' U X 1 42.706 29.395 48.257 1.00 0.00
ATOM 6 N1 U X 1 43.857 30.305 47.703 1.00 0.00
ATOM 7 C6 U X 1 45.057 29.857 47.308 1.00 0.00
ATOM 8 C5 U X 1 46.025 30.676 46.763 1.00 0.00
ATOM 9 C4 U X 1 45.720 32.110 46.702 1.00 0.00
ATOM 10 O4 U X 1 46.444 32.975 46.256 1.00 0.00
in a loop:
for i in *; do rna_add_chain.py -c A $i > ../struc/${i}; done
rna_add_chain.py¶
usage: rna_add_chain.py [-h] [-c CHAIN] file
- file¶
file
- -h, --help¶
show this help message and exit
- -c <chain>, --chain <chain>¶
a new chain, e.g. A
Measure distance between atoms¶
pdbs_measure_atom_dists.py¶
This is a quick and dirty method of comparison two RNA structures (stored in pdb files). It measures the distance between the relevan atoms (C4’) for nucleotides defined as “x” in the sequence alignment.
author: F. Stefaniak, modified by A. Zyla, supervision of mmagnus
usage: pdbs_measure_atom_dists.py [-h] [-v]
seqid1 seqid2 alignfn pdbfn1 pdbfn2
- seqid1¶
seq1 id in the alignemnt
- seqid2¶
seq2 id in the alignemnt
- alignfn¶
alignemnt in the Fasta format
- pdbfn1¶
pdb file1
- pdbfn2¶
pdb file2
- -h, --help¶
show this help message and exit
- -v, --verbose¶
increase output verbosity
This is a quick and dirty method of comparison two RNA structures (stored in pdb files). It measures the distance between the relevan atoms (C4’) for nucleotides defined as “x” in the sequence alignment.
author: F. Stefaniak, modified by A. Zyla, supervision of mmagnus
- rna_tools.tools.pdbs_measure_atom_dists.pdbs_measure_atom_dists.find_core(seq_with_gaps1, seq_with_gaps2)[source]¶
.
- Parameters:
seq_with_gaps1 (str) – a sequence 1 from the alignment
seq_with_gaps1 – a sequence 2 from the alignment
Usage:
>>> find_core('GUUCAG-------------------UGAC-', 'CUUCGCAGCCAUUGCACUCCGGCUGCGAUG') 'xxxxxx-------------------xxxx-'
- Returns:
core=”xxxxxx——————-xxxx-”
- rna_tools.tools.pdbs_measure_atom_dists.pdbs_measure_atom_dists.get_seq(alignfn, seqid)[source]¶
Get seq from an alignment with gaps.
Usage:
>>> get_seq('test_data/ALN_OBJ1_OBJ2.fa', 'obj1') SeqRecord(seq=SeqRecord(seq=Seq('GUUCAG-------------------UGAC-', SingleLetterAlphabet()), id='obj1', name='obj1', description='obj1', dbxrefs=[]), id='<unknown id>', name='<unknown name>', description='<unknown description>', dbxrefs=[])
- Returns:
SeqRecord