rna-tools & PyMOL [PyMOL4RNA]¶
See also https://github.com/mmagnus/rna-tools/tree/master/rna_tools/tools/PyMOL4RNA
Inspect structure¶
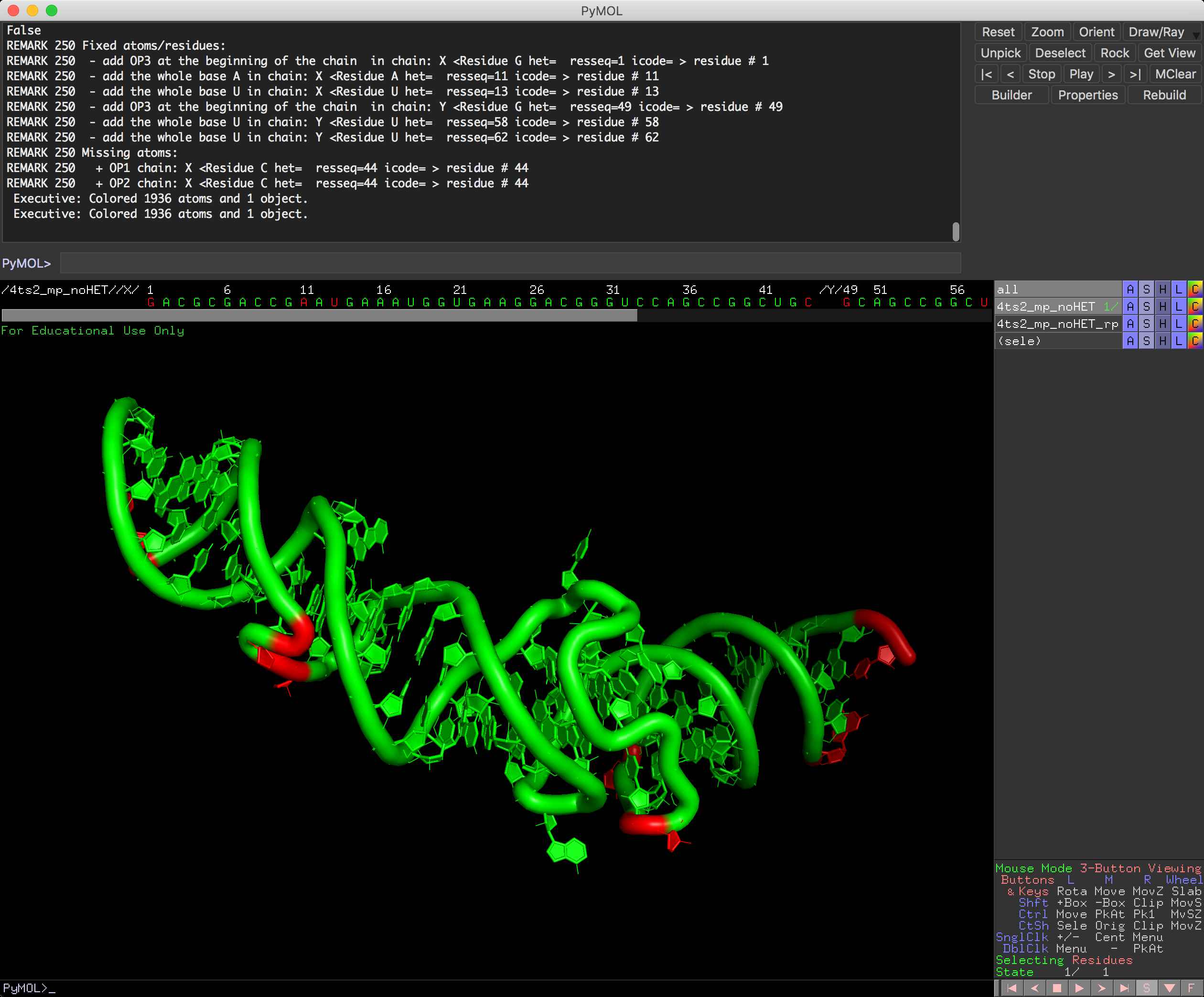
There is new `--inspect` function for rna_pdb_tools.py that tells you what are missing atoms in your structure file.
You can run it also from PyMOL, so in here you can see missing atoms directly in PyMOL (the object comes green and the residues missing atoms comes in red).
PyMOL:
inspect <object name>
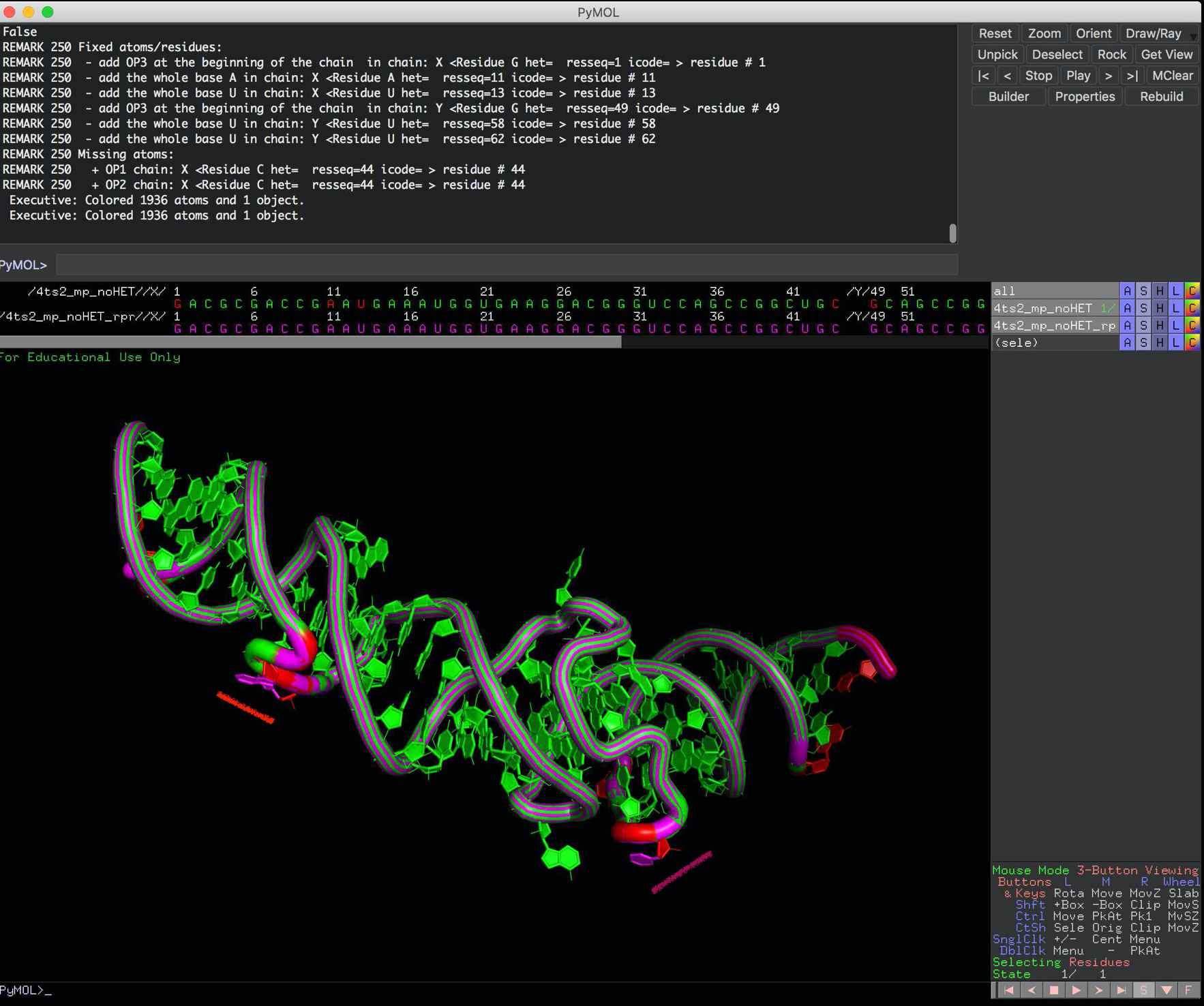
and here you can compare the input file vs file after --rpr (of rna_pdb_tools.py) to see what was rebuilt (in pink).
Show base pair edges¶
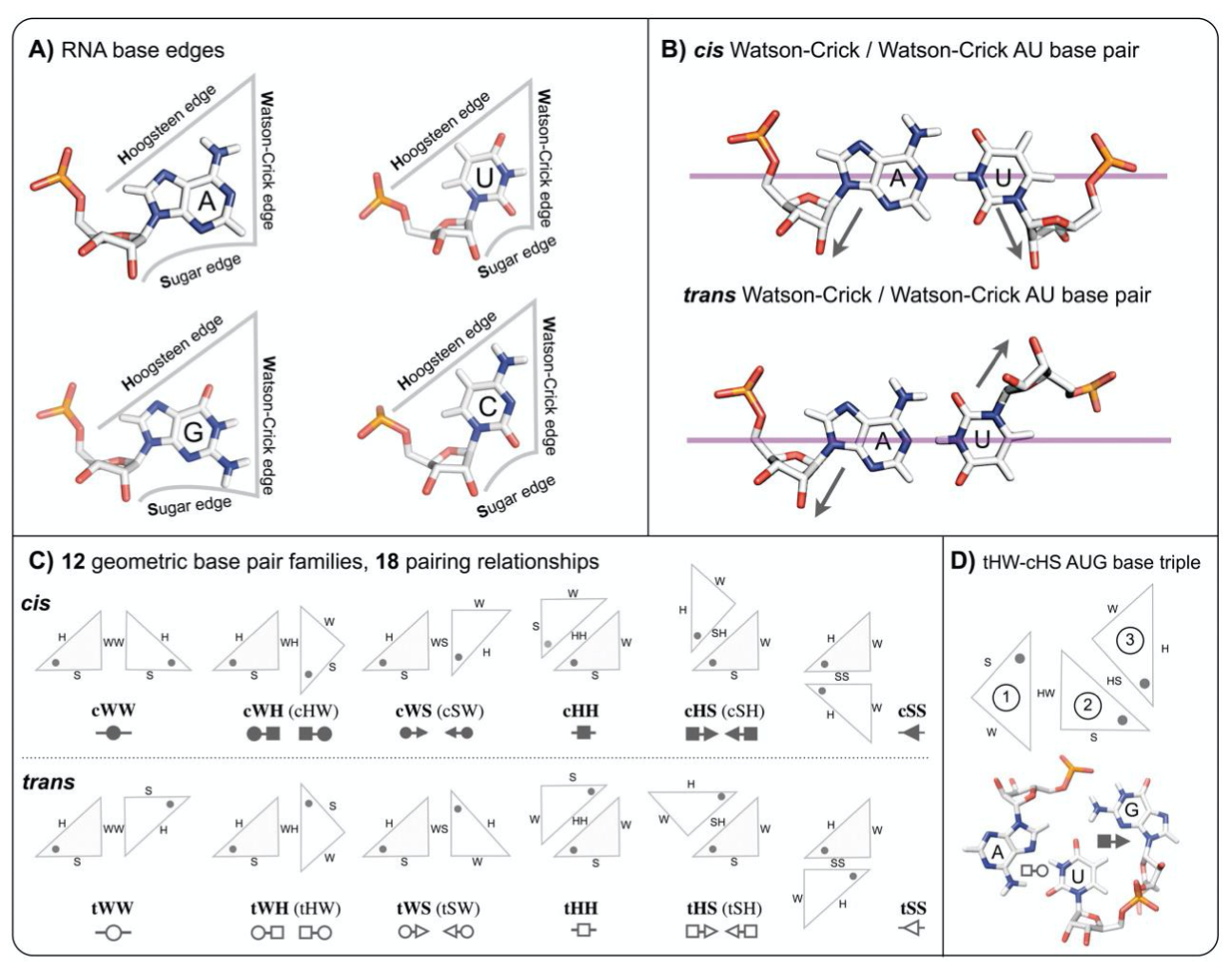
Leontis/Westhof classification of base pairings. (A) RNA bases - adenine (A), cytosine (C), guanine (G) and uracil (U) - involve one of three distinct edges: the Watson– Crick (W) edge, the Hoogsteen (H) edge, and the Sugar (S) edge. (B) Each pair of can interact in either cis or trans orientations with respect to the glycosidic bonds. (C) For these reasons, all base pairs can be grouped into twelve geometric base pair families and eighteen pairing relationships (bases are represented as triangles). Each pair is represented by a symbol that can be used in a secondary structure and a tertiary structure diagrams. Filled symbols mean cis base pair configuration, and open symbols, trans base pair. (D) Interestingly, bases can form triples and they have own classification devised by Leontis and coworkers (Creative Commons License) (A. S. Abu Almakarem, A. I. Petrov, J. Stombaugh, C. L. Zirbel, and N. B. Leontis, “Comprehensive survey and geometric classification of base triples in RNA structures.,” Nucleic Acids Research, vol. 40, no. 4, pp. 1407–1423, Feb. 2012.)
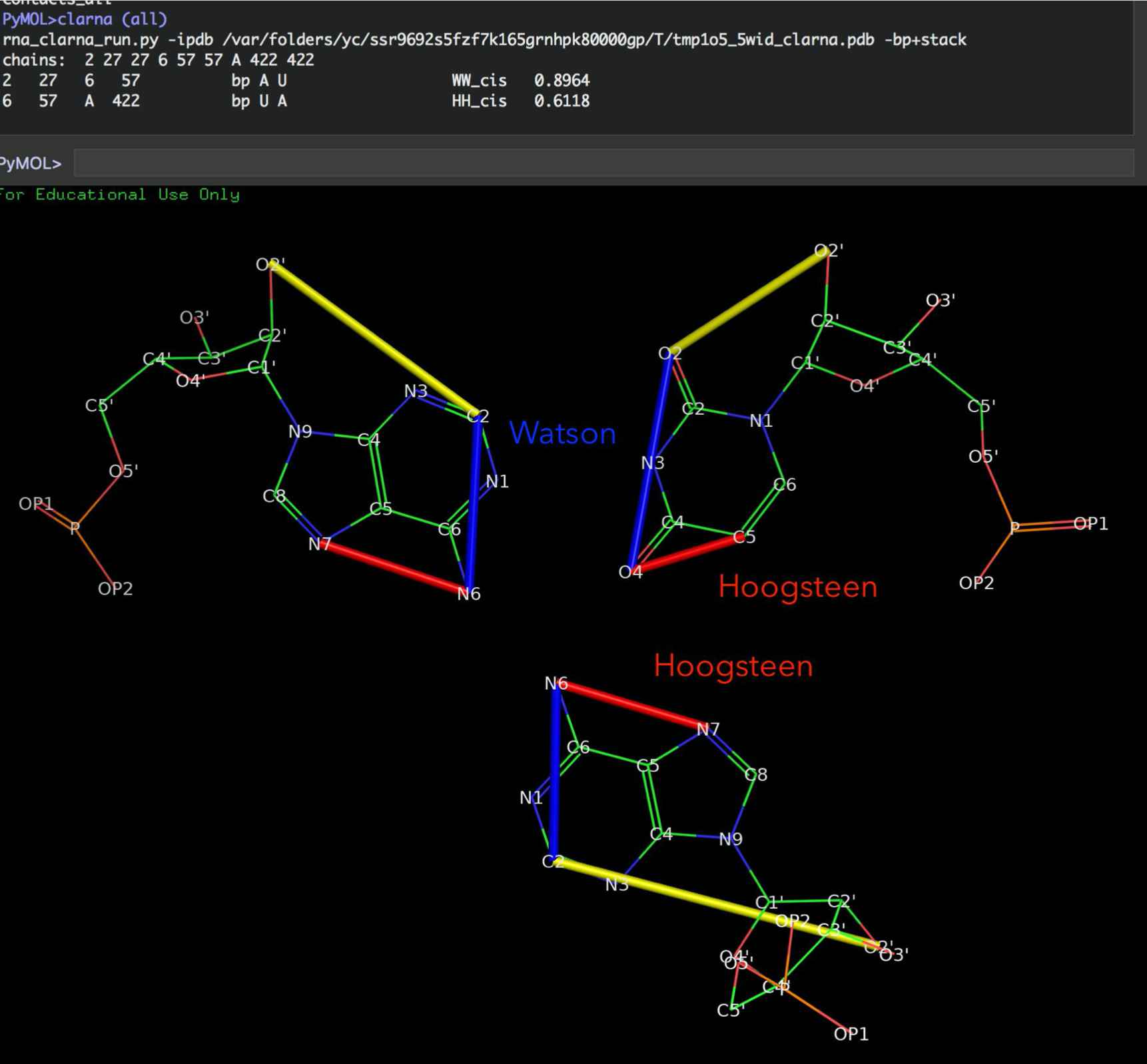
Warning
This PyMOL function is very simple and works on standard atom naming, so to make sure that everything will work, get your structure in the RNA Puzzle ready format, See https://rna-tools.readthedocs.io/en/latest/main.html#get-rnapuzzle-ready
Type:
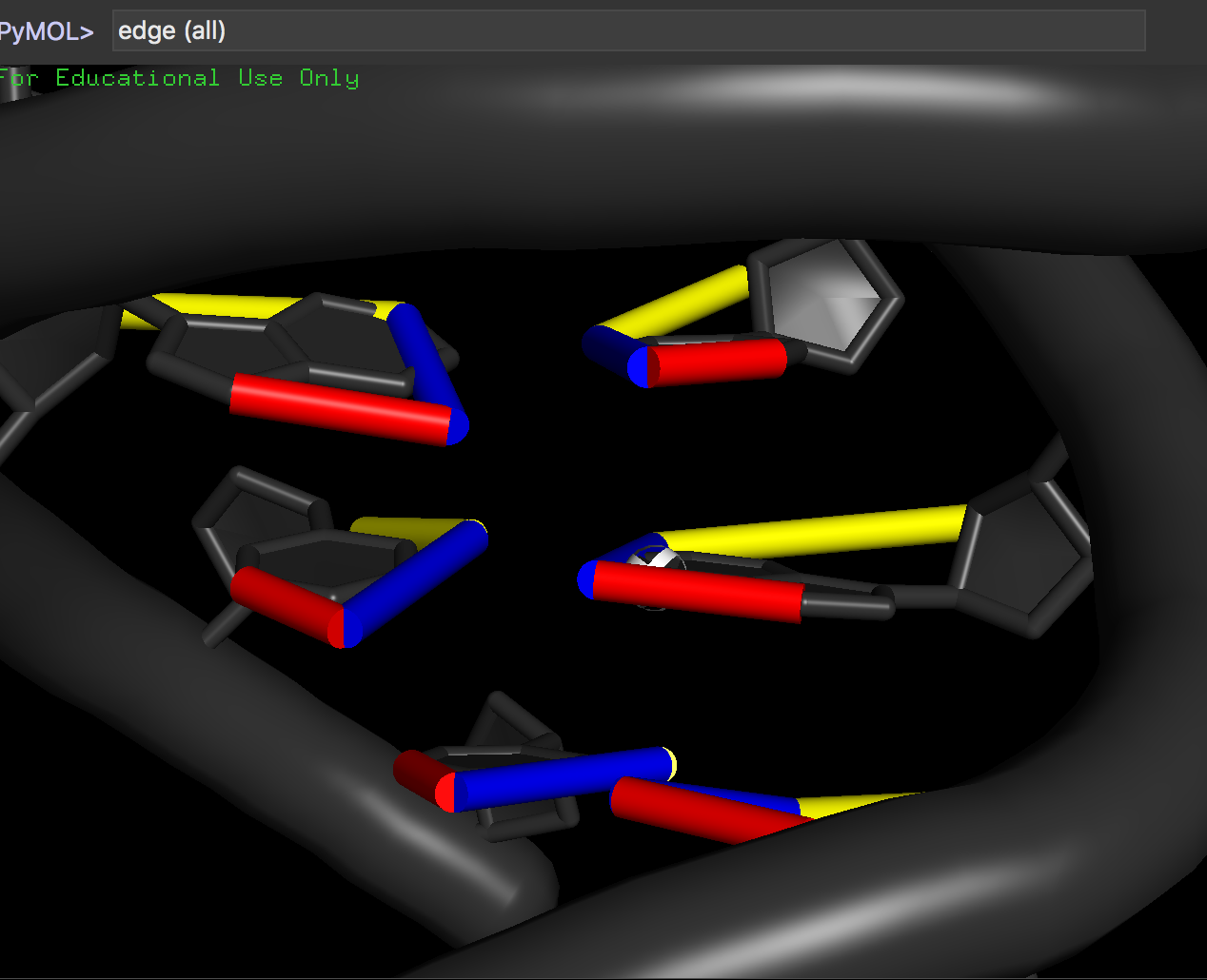
edges (<selection>)
e.g.,:
edges (all)
to show the edges drawn on bases.
PyMOL4Spliceosome: all spliceosome structures in one PyMOL session¶
Download one PyMOL session with structures for each step.
See own project https://github.com/mmagnus/PyMOL4Spliceosome
PyMOL: Color by conservation¶
Show conserved regions of proteins in PyMOL.
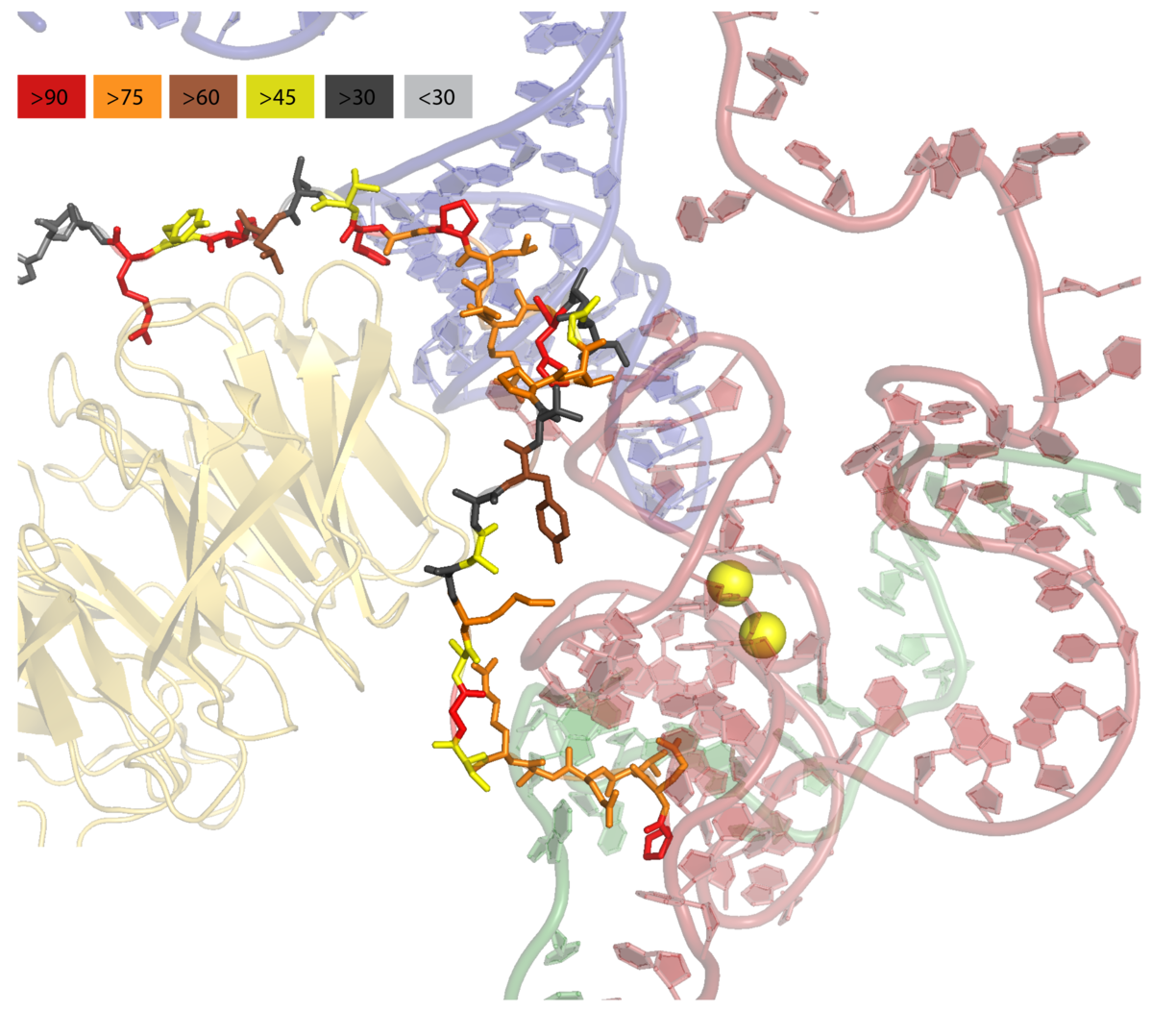
See more: https://github.com/mmagnus/rna-tools/tree/master/rna_tools/tools/pymol_color_by_conserv
PyMOL Drawing¶
- rna_tools.tools.pymol_drawing.pymol_drawing.draw_circle(x, y, z, r=8.0, cr=1.0, cg=0.4, cb=0.8, w=2.0)[source]¶
Create a CGO circle
- PARAMS
- x, y, z
X, Y and Z coordinates of the origin
- r
Radius of the circle
- cr, cg, cb
Color triplet, [r,g,b] where r,g,b are all [0.0,1.0].
- w
Line width of the circle
- RETURNS
the CGO object (it also loads it into PyMOL, too).
- rna_tools.tools.pymol_drawing.pymol_drawing.draw_circle_selection(selName, r=None, cr=1.0, cg=0.4, cb=0.8, w=2.0)[source]¶
circleSelection – draws a cgo circle around a given selection or object
- PARAMS
- selName
Name of the thing to encircle.
- r
Radius of circle. DEFAULT: This cript automatically defines the radius for you. If you select one atom and the resultant circle is too small, then you can override the script’s calculation of r and specify your own.
- cr, cg, cb
red, green and blue coloring, each a value in the range [0.0, 1.0]
- RETURNS
The circle object.
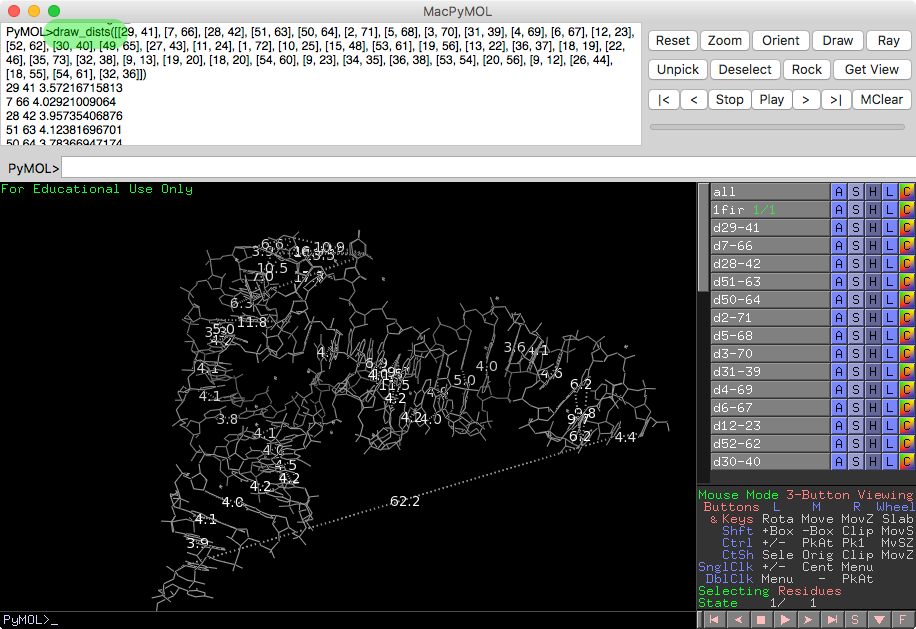
- rna_tools.tools.pymol_drawing.pymol_drawing.draw_dist(54.729, 28.9375, 41.421, 55.342, 35.3605, 42.745)[source]¶
Install PyMOL plugin to view the interactions with PyMOL:
run <path>rna-tools/tools/pymol_drawing/pymol_dists.py
and type:
draw_dists([[29, 41], [7, 66], [28, 42], [51, 63], [50, 64], [2, 71], [5, 68], [3, 70], [31, 39], [4, 69], [6, 67], [12, 23], [52, 62], [30, 40], [49, 65], [27, 43], [11, 24], [1, 72], [10, 25], [15, 48], [53, 61], [19, 56], [13, 22], [36, 37], [18, 19], [22, 46], [35, 73], [32, 38], [9, 13], [19, 20], [18, 20], [54, 60], [9, 23], [34, 35], [36, 38], [53, 54], [20, 56], [9, 12], [26, 44], [18, 55], [54, 61], [32, 36]])
Install¶
After you install rna-tools, run these two lines your terminal:
$ echo "sys.path.append('`rna_tools_which.py --site`')" >> ~/.pymolrc
$ echo "PyMOL4RNA_PATH='`rna_tools_which.py`/tools/PyMOL4RNA/PyMOL4RNA.py'; cmd.run(PyMOL4RNA_PATH)" >> ~/.pymolrc
so something like this will be added to your ~/.pymolrc:
sys.path.append('/Users/magnus/work/src/rna-tools')
PyMOL4RNA_PATH='/Users/magnus/work/src/rna-tools/rna_tools/tools/PyMOL4RNA/PyMOL4RNA.py'; cmd.run(PyMOL4RNA_PATH)
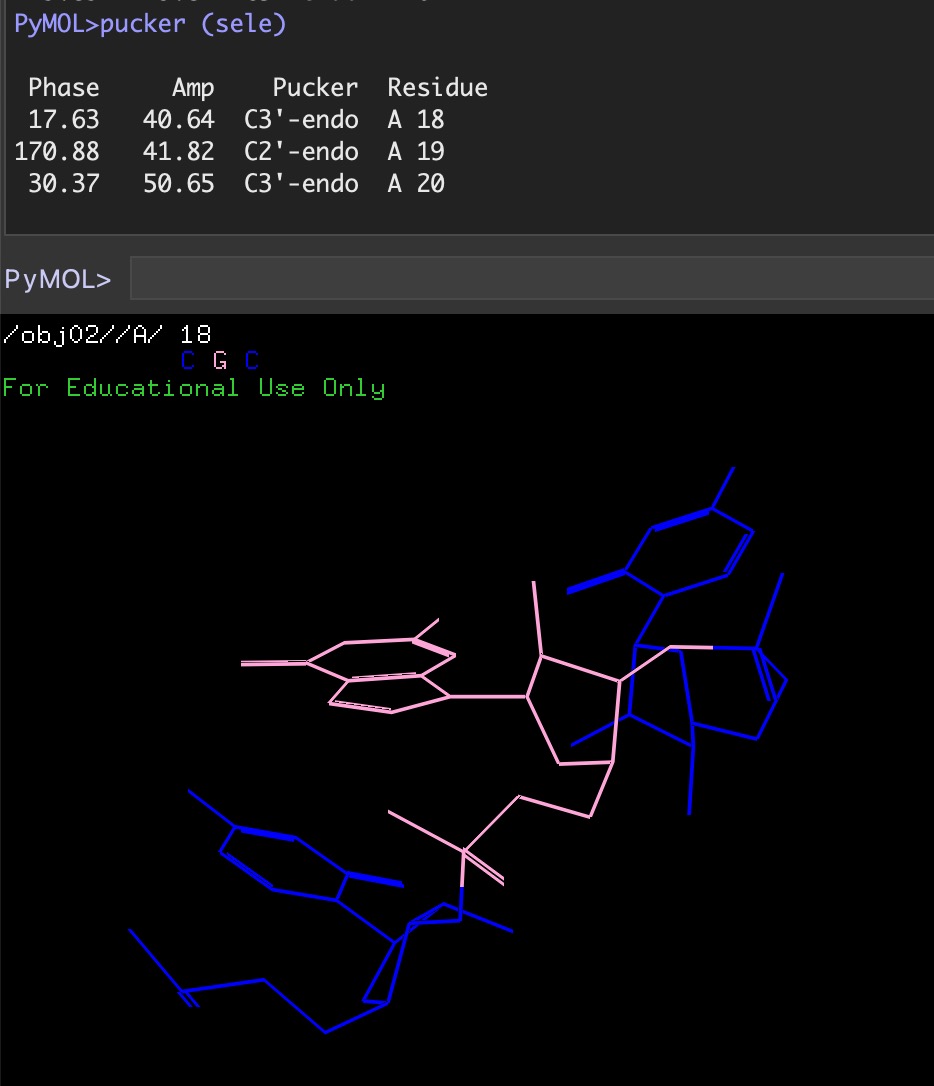
Libs: Pucker analysis¶
pucker.py is a PyMol script that returns the sugar pucker information (phase, amplitude, pucker) for a given selection.
This script uses its own dihedral calculation scheme rather than the get_dihedral command. Thus, it is lightning fast!
If a selection does not contain any ribose sugars then an error message is returned.
Author: Sean Law (Institute: University of Michigan) adapted for Python 3 by magnus <mmagnus@fas.harvard.edu>.
Install, add run <path>/rna-tools/rna_tools/tools/PyMOL4RNA/libs/pucker.py to your ~/.pymolrc
Source: <https://pymolwiki.org/index.php/Pucker>